DDBJ データ解析チャレンジ
DDBJ データ解析チャレンジでは、DDBJ 保有のビッグデータである塩基配列公開データベースを用いて、チャレンジ課題の機械学習モデルの精度を競います。
構築モデルは京都大学のビッグデータ大学に投稿します。
参加者がビッグデータ解析の経験を積める様に、学生やライフサイエンス専門外の方にも、遺伝研スーパーコンピュータの計算機環境を提供します。
初日には参加者向けにキックオフ講習会を行い、課題やスパコンの使い方を説明します。
DDBJ データ解析チャレンジ 2016
| 日程 | 内容 |
|---|---|
| 2016年6月27日(月) | キックオフ講習会 参加受付開始、遺伝研スパコン期間利用申請、OSSインストール申請受付開始 |
| 2016年7月6日(水) | 課題投稿の受付開始、キックオフ講習会 |
| 2016年8月21日(日) | 遺伝研スパコン期間利用申請、OSSインストール申請受付の締切 |
| 2016年8月31日(水) | 課題投稿の締切 |
| 2016年9月30日(金) | 結果発表 |
チャレンジ課題
DNA配列からクロマチン特徴を予測します。DDBJ Sequence Read Archive(DDBJ SRA)からクロマチン特徴を注釈したデータベースに「ChIP-Atlas(九大沖博士、DBCLS大田博士)」があります。ChIP-Atlasデータベースに未掲載の生物種を対象として、DNA配列がクロマチン特徴領域か否かを予測します。
- 背景と意義
- ゲノム研究分野では、GWAS解析やQTL解析により疾患・病害リスクとなるDNA多型の同定が進められています。DNA多型の疾患・病害リスクの解釈には、オープンクロマチン領域情報、ヒストン修飾情報、転写因子結合部位情報などの遺伝子発現のオンオフに関わるクロマチン特徴情報が重要です。クロマチン特徴は組織条件が変わるとオンオフが変わります。一方、近年ゲノム機能解析データの蓄積や計算機性能の向上により、DNA配列からクロマチン特徴を組織条件を含めてモデル化を行い、全条件のクロマチン特徴の一挙予測が可能になりました。全条件モデルを組み合わせると、疾患・病害リスクと関連するDNA多型を数多くの候補の中から絞り込む事が可能になります。 DDBJでは、高速DNAシークエンサ由来のビッグデータDDBJ SRAを提供しています。SRAを再解析したクロマチン特徴注釈データベース「ChIP-Atlas」を使えば、大規模なクロマチン特徴予測モデルの構築が可能になります。DNA多型候補の絞込み精度は、土台である遺伝子発現オンオフ特徴(=本チャレンジではクロマチン特徴)の精度に依存する為、高精度のクロマチン特徴予測モデルの構築が必要です。本チャレンジではChIP-Atlas注釈情報とDNA配列を用いて、高精度クロマチン特徴予測モデルの構築を目指します。
- データ説明と予測課題
- 課題データは、CellTypeクラスと Antigenクラスの組合わせ8条件で構成されています。8条件の訓練データを用いてモデルを構築し、テスト配列の予測結果を投稿して下さい。課題の8条件は、AntigenクラスはDNase-seq,Histone,RNA polymerase,TFs(Transcriptional factors)の4条件を含めて構成しています。生物種はChIP-Atlasデータベースに未掲載のモデル植物シロイヌナズナです。DNA配列ゲノム版は、TAIR10です。
- -----------------------------------
入力訓練データ :60,000 DNA配列
入力テストデータ:10,000 DNA配列
出力訓練データ :8条件の正解(真偽)
----------------------------------- - <入力>
1配列は、対象生物のゲノム上の200塩基(200個のACGTの並び)フラグメントで構成されています。
1配列は、01コードで保存しており1行800データです。[例:AATGC…=10001000000100100100…]
対応コード:A=1000,C=0100,G=0010,T=0001, その他の例外=0000
<出力>
出力訓練データ(正解データ)は01コードです。1が真で、DNA配列はクロマチン特徴領域を含む。0が偽でクロマチン特徴領域を含まない、に相当します。
<課題>
入力テストデータの予測結果として、1万行8列で真の予測確率をビッグデータ大学に投稿して下さい。(列間区切はスペース) - チャレンジ効果
- 課題のクロマチン特徴予測モデルが高精度になれば、遺伝子発現オンオフに関わる(即ち疾患・病害リスクとなる)DNA多型の絞込み精度向上に繋がります。構築したシロイヌナズナのクロマチン特徴予測モデルは、他の植物へ応用可能な為に植物研究コミュニティに貢献する事が出来ます。
表彰
| 最優秀賞 1st Prize of DDBJ Challenge Awards 2016 |
株式会社 情報数理バイオ 研究開発部 ライフサイエンスグループ 望月 正弘 |
| 優秀賞 2nd Prize of DDBJ Challenge Awards 2016 |
国立研究開発法人 理化学研究所 情報基盤センター バイオインフォマティクス研究開発ユニット 松本 拡高(代表)、尾崎 遼(チームとして2名で参加) |
| 優良賞 3rd Prize of DDBJ Challenge Awards 2016 |
ビッツ株式会社 岡山 利次 |
| 学生賞 Student Prize of DDBJ Challenge Awards 2016 |
東京大学大学院 情報理工学系研究科 修士課程1年 加藤 卓也 |
今回「DNA配列からのクロマチン特徴予測」のチャレンジ課題について、38名が参加して、延べ360回のモデル投稿がありました。
最優秀賞モデルの概要
望月氏のモデルは、Extremely Randomized Trees(ERT, 参考文献1) と Convolutional Neural Network(CNN, 参考文献2) の2種類の分類器を基盤として、Stacked Generalization(Stacking, 参考文献3) アンサンブル学習法で精度向上を図っています。
特徴量は、チャレンジのクエリ配列だけでなく外部特徴量(ゲノム座標、遺伝子注釈情報)を組み入れています。1つ目のERTモデルはゲノム座標が特徴量でn(配列数) x m(染色体数)の行列です。ゲノム座標はクエリ配列をシロイヌナズナTAIR10ゲノム参照配列(参考文献 4)にアライメントして得ます。このERTモデルをGenomic Coordinates Based Model(GCBM)とします。
2つ目のCNNモデルの特徴量は、クエリ配列と遺伝子注釈情報(TAIRからGFFファイルをダウンロード)です。Figure 1の様にforward/reverse strand別で遺伝子注釈情報を組み込んでいます。遺伝子注釈情報は定量値で、定義は次式になります。

変数rは減衰率で、変数dは遺伝子の1塩基目からの距離です。変数rが0なら特徴量は1になり、遺伝子中に対象塩基が含まれる事を表します。変数rが0より大きい時は、遺伝子開始塩基からの勾配値が与えられます。このCNNモデルをGene Annotated Sequences Based Model(GASBM)とします。
Figure1: Structure of the neural network of GASBM
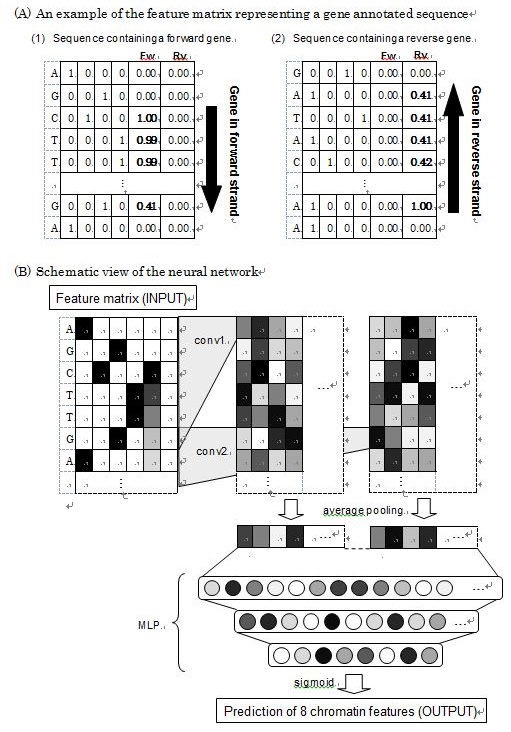
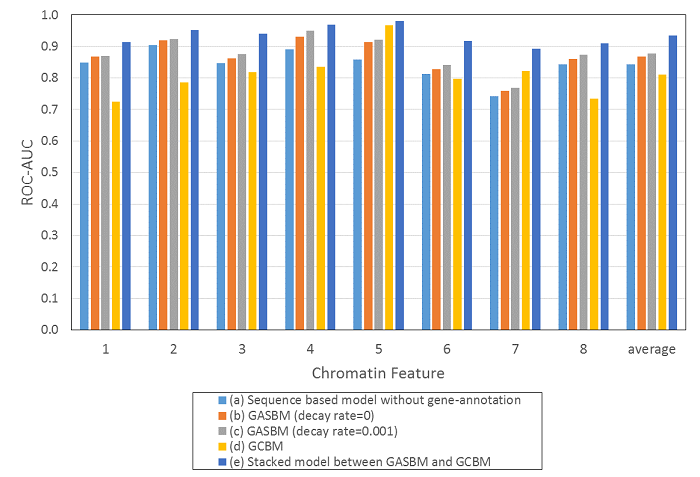
Figure2はベンチマーク結果です。ERT-GCBMモデルとCNN-GASBMモデルのパラメータ値などの詳細は、他の受賞者モデルも含めた全体解説(データクレンジング、特徴量選択、モデル訓練と予測法)と共に、報告論文で公開する予定です。
Figure2: Benchmark result of models
Reference
- [1] Geurts, P., Ernst, D. & Wehenkel, L. Extremely randomized trees. Machine Learning 63, 3-42 (2006).
- [2] LeCun, Yann, et al. “Gradient-based learning applied to document recognition.” Proceedings of the IEEE 86.11 (1998): 2278-2324.
- [3] Wolpert, D. H. Stacked generalization. Neural Networks 5, 241-259 (1992).
- [4] https://www.arabidopsis.org/
受賞モデルの精度結果
| DDBJ Challenge Award |
AUC | Model Design | Tool Version |
|---|---|---|---|
| 1st Prize | 0.94564 | 2 Classifiers(Extremely Randomized Trees, CNN) Ensemble Learning(Stacking) *External Data(Genomic Position, Gene Structure Annotation) |
python=3.5 scikit-learn=0.17.1 chainer=1.10.0 |
| 2nd Prize | 0.89859 | 2 Classifiers(CNN, Product of Genomic Distance Decay Parameter and Nearest Training Data Output) Ensemble Learning(Averaged) *External Data(Genomic Position) |
julia=0.4.6 python=2.7.10 skflow(tensorflow=0.8.0) |
| 3rd Prize | 0.85428 | 7 Classifiers(Naive Bayes for Multivariate Bernoulli Models, Logistic Regression, Random Forest, Gradient Boosting, Extremely Randomized Trees, eXtreme Gradient Boosting, CNN) Ensemble Learning (Stacking) |
python=2.7.11 numpy=1.10.4 scikit-learn=0.17 chainer=1.11.0 xgboost=0.4a30 |
| Student Prize | 0.84318 | 3 Classifiers(LeNet like CNN, DeepBind like CNN, Variable filter DeepBind like CNN) Ensemble Learning(Soft Voting) |
python=2.7 lasagne=0.2.dev1 |
論文
- DDBJ Data Analysis Challenge: a machine learning competition to predict Arabidopsis chromatin feature annotations from DNA sequences.
Kaminuma E, Baba Y, Mochizuki M, Matsumoto H, Ozaki H, Okayama T, Kato T, Oki S, Fujisawa T, Nakamura Y et al
Genes Genet Syst 2020 Mar 26;():. Pubmed: 32213716
DDBJデータ解析チャレンジ Q&A
- DDBJデータ解析チャレンジの内容や結果は論文に投稿できますか?
- DDBJチャレンジ報告論文をジャーナルに投稿予定です。上位入賞者の方には、共著者としてご参加いただきます。詳しくは チャレンジ賞 を参照ください。
- 自分の PC に機械学習のためのプログラミング言語の開発環境を準備した方がいいですか?
- NIG スパコンには、R, MATLAB, Python などの機械学習ツールがインストールされていますので、NIG スパコンにログインして作業を行う場合は、ご自身のPC内に開発環境を作成する必要はありません。(NIG スパコンを利用する場合はアカウント申請が必要です。) ローカルPC で作業を行う場合には、ご自身で環境整備をして頂く必要があります。ローカルPCには期間中のみ無料利用できるMATLABをインストール可能です。
- チャレンジで利用した遺伝研スパコンのアカウントは、チャレンジ終了後どうなりますか?
- 利用終了後の9月1日から、アカウントは使用できなくなります。 ライフサイエンス研究の為に使用したい方は、アカウント継続申請を こちら からリクエストしてください。 継続申請フォームには下記を入力してください。 お問い合わせ種別:「その他」を選択してください。 お問い合わせ件名:「チャレンジアカウントの継続申請」を入力してください。
- 遺伝研スパコンを必ず使う必要がありますか?
- 遺伝研スパコンを使っても、使わなくてもどちらでも結構です。
- 遺伝研スパコンの利用申請時に、フリーランス(または会社員)なので責任者を記入できないです。
- フリーランスの方は経歴情報を、会社員の方はご所属の会社情報を、遺伝研スパコンお問い合わせ窓口 までご連絡ください。
- Tutorialでは遺伝研スパコンのメモリ要求は24GBです。自分のプログラムではメモリエラーになりました。
- TutorialはMATLAB線形判別分析用のメモリ設定になっています。最大64GBまで設定可能 (-l mem_req=64G , -l s_vmem=64G) なので、プログラム毎に最適な値を指定してください。64GBを超えた値を設定すると、core dump 出力でジョブは強制終了しますので御注意ください。
- 配列データに0000コードを発見しました。また0000コードのみでクロマチン情報有の配列がありました。
- ゲノム領域の一部で塩基未決定の場合があり、課題では0000コードに設定しています。課題中の未決定塩基のみの配列は、あるピーク領域中の断片として、そのクロマチン情報が出力値に入っています。参加の皆様自身で、良い対処策を御考案下さい。
- challenge.q使用率が100%になっていて、使えなくて困っています。
- challenge.qノード数を16に変更しました。またリソースの占有を防ぐ為に、challenge.qの同時使用可能スロット数を150に変更しました。
- DDBJ-challenge.mat以外の外部データ利用は許可されていますか?
- 外部データ利用を許可していますが、下記の様に条件を設定します。
パラメータ(注1)の外部利用=可
転移学習用Pretrained Modelの利用=可(注2)
(注1)=馬場博士スライドp.15参照
(注2)=不正を防ぐ為に、外部入力配列は植物を禁止します - testデータを訓練に使用(semi-supervised learning)して良いですか?
- はい、良いです。
- Deep Learning用ライブラリを遺伝研スパコンへインストールするとエラーが出ます。
- UC Berkeley の Caffe, Preferred Networks社 Chainer のインストール方法を スパコンHP に掲載しましたのでご参照ください。
DDBJ チャレンジ実施委員会
- DDBJ Challenge Committee
- 神沼英里 : 国立遺伝学研究所 生命情報研究センター 助教 鹿島久嗣 : 京都大学大学院 情報学研究科 教授 高木利久 : 国立遺伝学研究所 生命情報研究センター 教授
本チャレンジは遺伝研所内倫理審査委員会(IRB)により倫理審査承認を受けております。
お問い合わせ:DDBJへのお問い合わせ の「講習会」からご連絡ください。