INSD has expanded its specifications to accept data submissions from large scale sequencing projects. For example, we have started accepting the sequence data from EST (Expressed Sequence Tags) projects into EST division since 1993. On 2002, to accept submissions of the draft genome and the meta-genome sequences, we have created the new category for WGS (Whole Genome Shotgun) data.
The left figure shows the relationship of the ratios of three categories. The right figure shows the numbers of bases.
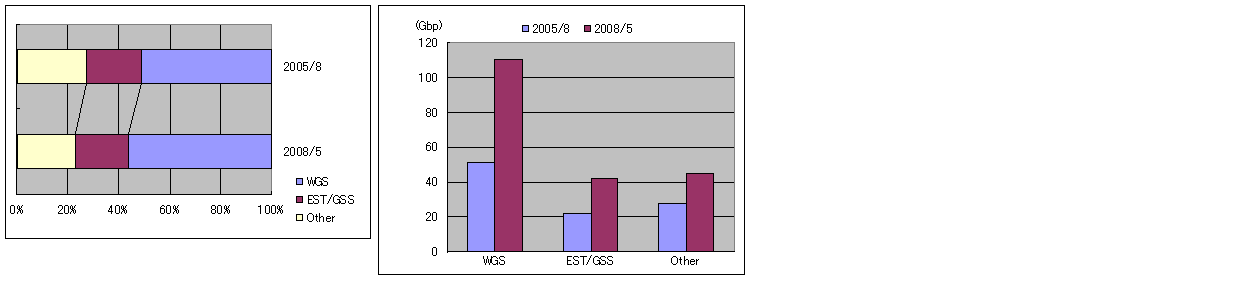
As you can see in the right figure, base counts of both WGS and EST/GSS (Genome Survey Sequences) data increased approximately two times during three years. The base count of WGS data has increased 100 G bases only past 6 years.
What is WGS data?
In general the term, WGS "Whole Genome Shotgun", originally means an approach to sequence genomic DNA molecules that are once fragmented into millions of pieces, which are sequenced and reassembled to produce a series of sequences,. The strategy was firstly adopted to sequence the complete genome of Haemophilus influenzae in 1995. In INSD, the large set of contigs or the finished sequences without annotation from the proceeding genome project can be accepted as WGS data.
Future improvement
Increasingly High-Throughput Sequencing (HTS) technology (also called the next generation sequencers) is gaining popularity. It will accelerate the increase ratio of sequence data submitted to DDBJ. For the vast quantities of future submissions, DDBJ has to improve our database systems.
NCBI has started making SRA (Short Read Archive) to collect the raw outputs of short piece data derived from the next-generation sequencer, such as 454, Solexa, SOLiD and so on. As well as EBI, DDBJ is planning to collaborate with SRA.
Related to the new sequencing technologies, INSD faces many requests for the deposition of assembled EST (or EST-like short piece of) sequences. Therefore we will prepare a new division, TSA (Transcriptome Shotgun Assembly), to accept assembled sequences derived from transcriptome projects.
The day of reaching to 300 G bases
In February 2008, NIH announced about the 1000 Genomes Project, and they already sequenced human genome data between 200 to 300 G bases in their count. Related to this kind of project, it is not enough for DDBJ to accept huge scale of nucleotide sequences.
Because research communities require not only the text based nucleotide sequences, but also the raw outputs of trace data for the sequences to investigate reliabilities of sequence data, evaluation of polymorphisms among individuals and so on. Trace Archive has accepted huge number of trace data registration, more than tens of terabytes (1013 order) in their total size. Last year, DDBJ started to accept a part of trace data from Japanese researchers for the first trial. It is supported by the Integrated Database Project.
Research analyses of large scale sequences will intend to the peta byte (1015 order) data. Considering with only the size of sequence data in INSD (i.e. excluding SRA and Trace Archive), the number of nucleotides is possible to be 400 G bases or more in one and a half year later.