Sequence Read Archive
データファイル
- ファイル名は英数字 [A-Z,a-z,0-9]、アンダースコア [_]、ハイフン [-] とドット [.] のみから構成され、空白文字、カッコ、句読点やシンボルを含まないこと。
- ペアリードの fastq は forward と reverse を別々のファイルにして、一つの Run に登録します。BAM ファイルは分割する必要はありません。
- データファイルは登録用ディレクトリの直下にアップロードしてください。
- サブディレクトリは作成しないでください。
- tar などでアーカイブしないでください。
- BAM ファイルは圧縮しないでください。
一般的な形式
fastq
Run の filetype は fastq を選択します。
fastq の形式。詳しくは NCBI のサイトをご覧ください。
- Quality value は phred 形式にしてください。オフセットはデフォルトで 33 (!) になります。64 (@) の場合は Run XML を編集して ascii_offset=”@” にしてください。
- DRA のメタデータ作成ウェブ画面は Technical read (アダプター、リンカー、バーコード配列) 記載に対応していません。記載する場合は Experiment XML を編集・登録してください (XML の記載例。
- ペアードリードは別々の fastq ファイルとして一つの Run に登録してください。ペアは標準形式のリード名から判定されます。
- リードの先頭は ‘@’ で始まっている必要があります。
- 塩基配列と Quality value は ‘+’ で始まる行で区切られている必要があります。
- fastq ファイルは gzip で圧縮してください。
BAM
BAM は Sequence Alignment/Map (SAM) ファイル形式のバイナリー圧縮です。 BAM ファイルは samtools でテキスト形式である SAM に変換することができます。
プライマリーデータとして unaligned read (アライメントされなかったリード) を含む BAM を登録することを推奨します。
SAM はリードの既知リファレンス配列へのアライメントに関する情報と生リードデータを含んだタブ区切りテキストファイルです。 SAM ファイルは二つの主要なセクション、ヘッダーとアライメントセクション、から構成されます。 ここでは「BAM ファイルの DRA への登録」という観点で SAM 形式について説明しています。 より詳細なファイルフォーマット仕様は samtoolsを参照してください。
SAM ヘッダーの例:
@HD VN:1.4 SO:coordinate
@SQ SN:CHROMOSOME_I LN:15072423
UR:ftp://ftp.ncbi.nlm.nih.gov/genbank/genomes/Eukaryotes/invertebrates/Caenorhabditis_elegans/
WBcel215/Primary_Assembly/assembled_chromosomes/FASTA/chrI.fa.gz AS:ce10
SP:Caenorhabditis elegans
@SQ SN:CHROMOSOME_II LN:15279345
UR:ftp://ftp.ncbi.nlm.nih.gov/genbank/genomes/Eukaryotes/invertebrates/Caenorhabditis_elegans/
WBcel215/Primary_Assembly/assembled_chromosomes/FASTA/chrII.fa.gz AS:ce10
SP:Caenorhabditis elegans
@RG ID:1 PL:ILLUMINA LB:C_ele_05 DS:WGS of C elegans PG:BamIndexDecoder
@PG ID:bwa PN:bwa VN:0.5.10-tpx
SAM アライメントの例:
3658435 145 CHROMOSOME_I 1 0 100M CHROMOSOME_II 2716898 0
GCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCT
AAGCCT
@CCC?:CCCCC@CCCEC>AFDFDBEGHEAHCIGIHHGIGEGJGGIIIHFHIHGF@HGGIGJJJJJIJJJJJJJJJJJJJJJJJJJJJHHHHHFF
FFFCCC RG:Z:1 NH:i:1 NM:i:0
5482659 65 CHROMOSOME_I 1 0 100M CHROMOSOME_II 11954696 0
GCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCTAAGCCT
AAGCCT
CCCFFFFFHHGHGJJGIJHIJIJJJJJIJJJJJIJJGIJJJJJIIJIIJFJJJJJFIJJJJIIIIGIIJHHHHDEEFFFEEEEEDDDDCDCCCA
AA?CC: RG:Z:1 NH:i:1 NM:i:0
BAM ファイルの処理
ヘッダーとアライメントセクションは整合的である必要があります。 各アライメントリードの RNAME (リファレンス配列の名前、3フィールド目) はヘッダー中の SN タグ値 (例 CHROMOSOME_I) と一致している必要があります。 加えて、アライメントリードの任意フィールドである read group (RG:Z:) はヘッダー中の read group ID と一致している必要があります (例 1)。 また、2フィールド目の FLAG フィールドはデータに対して正しい値がセットされていることが重要です。 DRA ファイル処理では不正な FLAG 値を補正しますが、不正な値が多すぎる場合はエラーになります。 DRA はアライメントセクション中の任意かつ非標準のタグ/フィールド値は保持しませんが、ヘッダーセクション全体は保持されます。
SAM フォーマットはリードのリファレンスへの全体マッチを表すイコールサイン (=) の sequence フィールドでの使用、もしくは、アスタリスク (*) の sequence と quality 両フィールドでの使用を許容していますが、DRA ではこれらの値を認識しません。
予期せぬ表記方法で記されたペアリードは適切に認識されず不適切な SRA ファイル形成をもたらします(ペアリードがシングルフラグメントとして扱われてしまいます)。
例えば、リード名の後ろに :0 と :1 と付されたリードはペアのリード1と2として認識されません。
これらの表記方法が使用されている 合、:0 と :1 を削除して同じリード名を使ったほうがよいです。
特定のシークエンサから出力される標準的な表記方法は正しく認識されます。
例えば、Illumina のリード名に付された /1 と /2 は標準的な表記方法です。
ペアリードに対して SAM BAM flag に適切なビット値 (multi-segment template 1-bit、first segment 64-bit と last segment 128-bit) が設定されていない場合、もしくは、ペアリードが別々の bam ファイルに分かれている場合は適正な SRA ファイルが生成されません。
BAM ファイルの登録
Run にアライメントデータを登録する場合は、「BAM」、「INSDC, refseq アクセッション番号 OR リファレンス配列マルチ fasta、それから bam SN リファレンス名、との対応表」が必要です。1 Run に 1 bam ファイルを登録します。
Run ではなく Analysis に登録する場合対応表は不要ですが、アライメントされなかったリードを含めた bam を Run に登録することを強く推奨します。
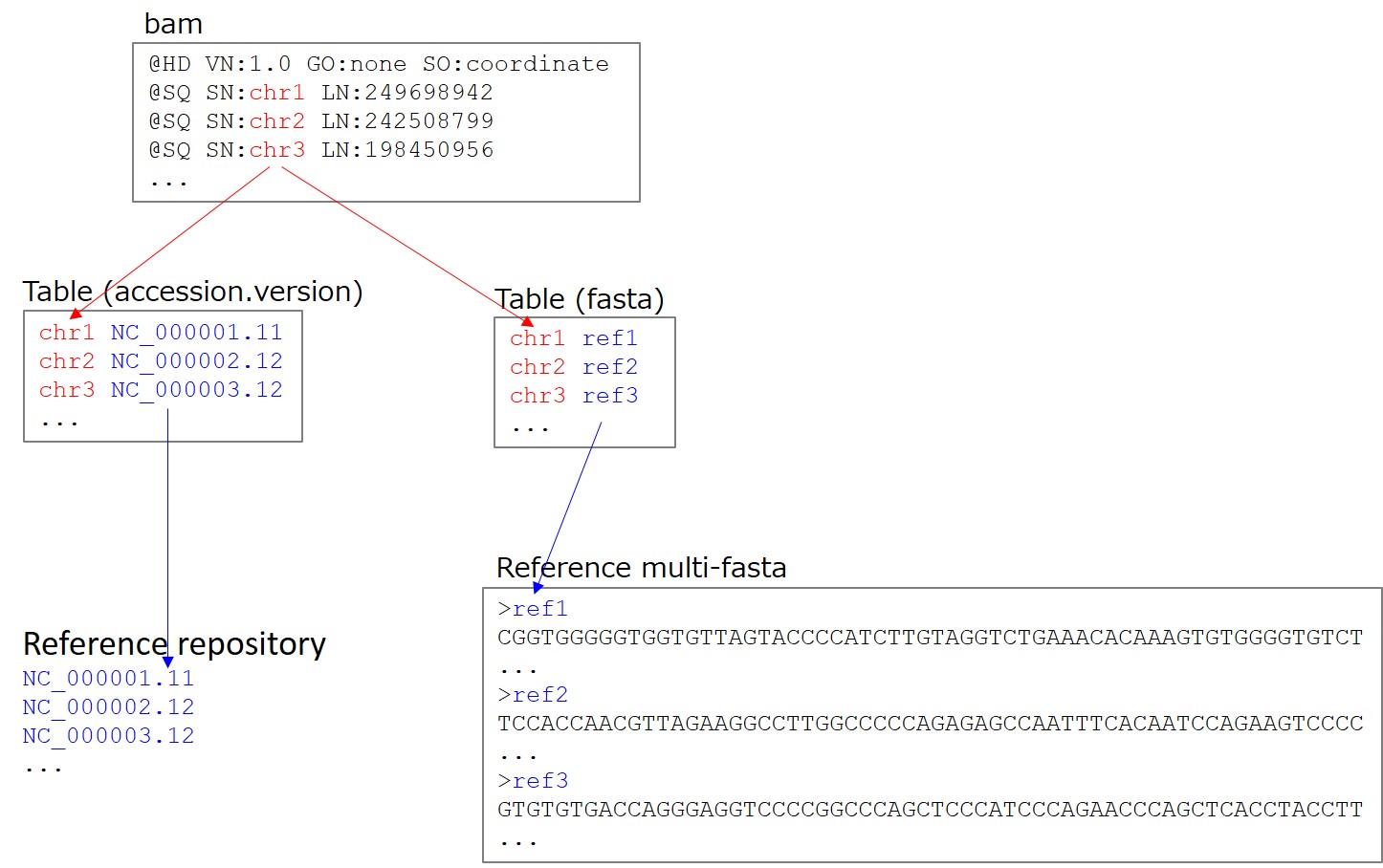
1. BAM
アライメントデータを BAM フォーマットで登録することができます。BAM ファイルは SAMtools と picard で読み込める形式になっている必要があります。圧縮していない BAM ファイルをアップロードしてください。
Run の File Type には “bam” を選択します。
2. リファレンスを INSDC/RefSeq アクセッション番号で指定
リファレンス配列が https://ftp.ncbi.nlm.nih.gov/sra/refseq/ にある場合、アクセッション番号.バージョン番号 (例 NC_000001.11) でリファレンスを参照することができます。 配列のバージョン番号は必須です。リファレンスゲノム配列のアクセッション番号は NCBI Assembly で検索することができます。
3. リファレンスをマルチ fasta で提供
リファレンス配列が https://ftp.ncbi.nlm.nih.gov/sra/refseq/ にない場合、リファレンス配列をマルチ fasta ファイルで提供します。真核生物のオルガネラ配列等短い配列は番号指定に対応していないケースがあります。 Run の File Type には “reference_fasta” を選択します。bam ヘッダーで定義されたリファレンスとマルチ fasta 中の配列は対応表を介して defline 中の配列名でリンクされます。 bam SQ 行 LN タグのリファレンス配列長とマルチ fasta 中の配列長が異なっている場合ワーニングになります。
4. INSDC/RefSeq アクセッション番号とマルチ fasta が混在するケース
一部のリファレンス配列が https://ftp.ncbi.nlm.nih.gov/sra/refseq/ にある場合、アクセッション.バージョン番号 (例 NC_000001.11) で一部のリファレンスを指定し、残りのリファレンス配列はマルチ fasta ファイルで提供します。混在しているケースでは、対応表にアクセッション.バージョン番号とマルチ fasta 中の defline 中の配列名を記載します。
5. SN-リファレンス配列の対応表
ご自分で独自に作成するファイルです。「BAM ファイルヘッダーの SQ 行中の SN 値」と「アクセッション番号 OR リファレンスマルチ fasta ファイル中の配列名」との対応関係をタブ区切りで記載します。 Run の File Type には “tab” を選択します。
BAM ファイルヘッダー
@HD VN:1.0 GO:none SO:coordinate
@SQ SN:chr1 LN:249698942
@SQ SN:chr2 LN:242508799
@SQ SN:chr3 LN:198450956
...
SN-リファレンス配列の対応表。例では SN:chr1 にリファレンス fasta ファイル中の配列 ref1 が対応。
chr1 ref1
chr2 ref2
chr3 ref3
...
リファレンスマルチ fasta。
>ref1
CGGTGGGGGTGGTGTTAGTACCCCATCTTGTAGGTCTGAAACACAAAGTGTGGGGTGTCT
...
>ref2
TCCACCAACGTTAGAAGGCCTTGGCCCCCAGAGAGCCAATTTCACAATCCAGAAGTCCCC
...
>ref3
GTGTGTGACCAGGGAGGTCCCCGGCCCAGCTCCCATCCCAGAACCCAGCTCACCTACCTT
...
SN-リファレンス配列の対応表。例では SN:chr1 に NC_000001.11 が対応。
chr1 NC_000001.11
chr2 NC_000002.12
chr3 NC_000003.12
...
プラットフォーム毎の説明
以下ではシークエンスプラットフォーム毎にデータファイル形式について説明しています。
454
454 からのシークエンスデータは fastq/bam ファイルで登録します。出力される sff ファイルは fastq/bam に変換したうえで登録してください。
Illumina
fastq/bam ファイルで登録します。
BGI-seq
fastq/bam ファイルで登録します。
SOLiD
fastq/bam ファイルで登録します。
Ion Torrent
fastq/bam ファイルで登録します。Ion Torrent から出力される bam ファイルは samtools で fastq に変換することができます。Converting BAM to fastq
Helicos Heliscope
quality value をすべて “14” として作成した fastq/bam ファイルを登録します。
Complete Genomics
fastq/bam ファイルで登録します。
Pacific Biosciences
HDF5
Pacific Biosciences は生データを格納するためにディレクトリ様構造を持つ HDF5 ファイルを使用しています。DRA は bas.h5 と bax.h5 両方のファイル形式での登録を受け付けています。RS II から出力されるデータは、1 の Run に *.bas.h5 1ファイルと *.bax.h5 3ファイルを登録します。ファイル名を変更しないでください。
Run に HDF5 以外のデータを含めないでください。
bam
以下の unaligned bam ファイルの登録をサポートしています。1 Run に 1 bamファイルを指定してください。 unaligned bamの場合、リファレンス配列や対応表の指定は必要ありません。
- *.subreads.bam
- *.ccs.bam
- *.reads.bam
fastq
fastq を Run の filetype で指定してください。
Oxford Nanopore
fastq/bam ファイルを登録します。
キャピラリシークエンサ
fastq/bam ファイルを登録します。
10x Genomics
10x Genomics のデータファイルについては GEA Single-cell submission guide を参照してください。
Analysis データファイル
Analysis への登録を推奨しているデータファイルについて説明します。
PacBio Base Modification Files
PacBio シークエンサーは塩基配列に加え、塩基のメチル化状態を解析することができ、そのデータを共有することは研究者コミュニティにとって重要です。例えば、修飾された塩基の正確な位置情報は DNA メチルトランスフェラーゼの特異性の解析に用いることができます。PacBio の解析ソフトウェア(RS_Modification_and_Motif_Analysis) はこれらの修飾塩基配列情報を抽出し、以下のいくつかのファイルに出力します。
- motif_summary.csv
- modifications.csv
- modifications.gff
- motifs.gff
この解析結果ファイル、原核生物については少なくとも motif_summary.csv ファイルを DRA Analysis として登録することは研究者コミュニティにとって貴重なデータになります。 Run に加え、これらのファイルを Sequence Annotation type Analysis としてご登録ください。サポートが必要な場合は DRA チームに連絡します。
NCBI guidelines of PacBio Base Modification Files
BioNano Whole Genome Map Files
BioNano はラベルの位置を染色体レベルで測定し、マッピングデータとして出力します。 マッピングデータはゲノムアセンブリ、構造バリアント検出、アセンブリの改善といった各種ゲノム解析に使われています。 次世代シークエンスデータと組み合わせ、より連続したハイブリッドゲノムアセンブリを得るために活用されています。 BioNano のマッピング・ハイブリッドアセンブリでは以下のファイル群が生成されます。
- CMAP
- BioNano の cmap ファイルはゲノムマップ中のラベル位置を測定した生データです。 cmap ファイルはタブ区切りテキストファイルでマップ上のラベル位置が開始・終了座標で記されています。
- COORD
- coord ファイルはハイブリッドアセンブリにおいてコンティグ座標と AGP ファイルを関連付けます。 coord ファイルは cmap の位置情報をリファレンスゲノムにマップしたものです(ギャップが両端に無い場合もあります)。 コンティグは、最も左端にある配列中の左端のラベルまで、最も右端にある配列の右端のラベルまで、トリミングされています。
- XMAP
- BioNano の xmap ファイルは二つのマップの比較結果です。 xmap ファイルはクエリの cmap とクエリ対象の cmap をアライメントし、比較した結果が記載されています。 xmap はタブ区切りテキストファイルで、マップの開始・終了座標、及び、マップにおけるラベル位置から構成されています。
- SMAP
- BioNano の smap ファイルはクエリの cmap とクエリ対象の cmap をアライメントし、検出された構造バリアント (SV) が記載されています。 smap はタブ区切りテキストファイルで SV の開始・終了座標、及び、マップにおける SV の位置から構成されています。
- BNX
- BioNano の bnx ファイルは染色体におけるラベル位置の測定結果、及び、チャンネル毎の Quality score の生データです。
最新のファイル仕様については Bionano Genomics - Software and Data Analysis Support Materials をご覧ください。
BioNano データをアセンブルに使用した場合、bnx ファイル、それから、de novo アセンブリに使用した EXP_REFINEFINAL1.cmap と coord ファイルを DRA Analysis として共有することは有益です。DRA では BioNano Genome Map データ用の analysis type と filetype を追加する予定ですが、それまでの間、analysis type “De Novo Assembly” と filetype “tsv” として BioNano データを登録してください (例 DRZ078181, DRZ078182)。
Antibiogram
薬剤感受性表(Antibiogram)は Analysis で受付けています。
メタデータエクセルで Analysis type は “Sequence Annotation” を選択し、Analysis シートの内容を埋めてください。薬剤感受性表はタブ区切りテキストファイルで登録します。BioProject/BioSample は登録済みのものを記入してください。
表の形式については NCBI BioSample の Antibiograms 関連のページをご参照ください。
NCBI BioSample Documentation
登録例