Sequence Read Archive
DRA 登録
- ヒトを対象とした研究データの登録について
- 研究対象者に由来するデータを DDBJ センターが運営するデータベースに登録する場合、研究対象者の尊厳及び人権は、適用されるべき法令、指針、ガイドライン、登録者が所属している機関の方針に従い、登録者の責任において保護されている必要があります。原則として、研究対象者を直接特定し得る情報はメタデータから取り除いてください。 ヒトを対象とした研究データを登録する場合は「ヒトを対象とした研究データの登録について」を熟読してください。
以下のデータについては、種類に応じたデータ登録が必要になりますので、専用ページをご覧ください。
登録の流れ
- DDBJ アカウントの取得
- 新規登録の作成
- データファイルのアップロード
- 登録ステータス
- BioProject の登録
- BioSample の登録
- Submission
- Experiment/Run の入力
- データファイルの検証処理
- アクセッション番号
DDBJ アカウントの取得
DDBJ アカウントを取得し、公開鍵をアカウントに登録します。
公開鍵がアカウントに未登録の状態で DRA 登録サイトにアクセスすると、以下のような警告が表示されます。 認証用公開鍵の登録に従って鍵を登録してください。
新規登録の作成
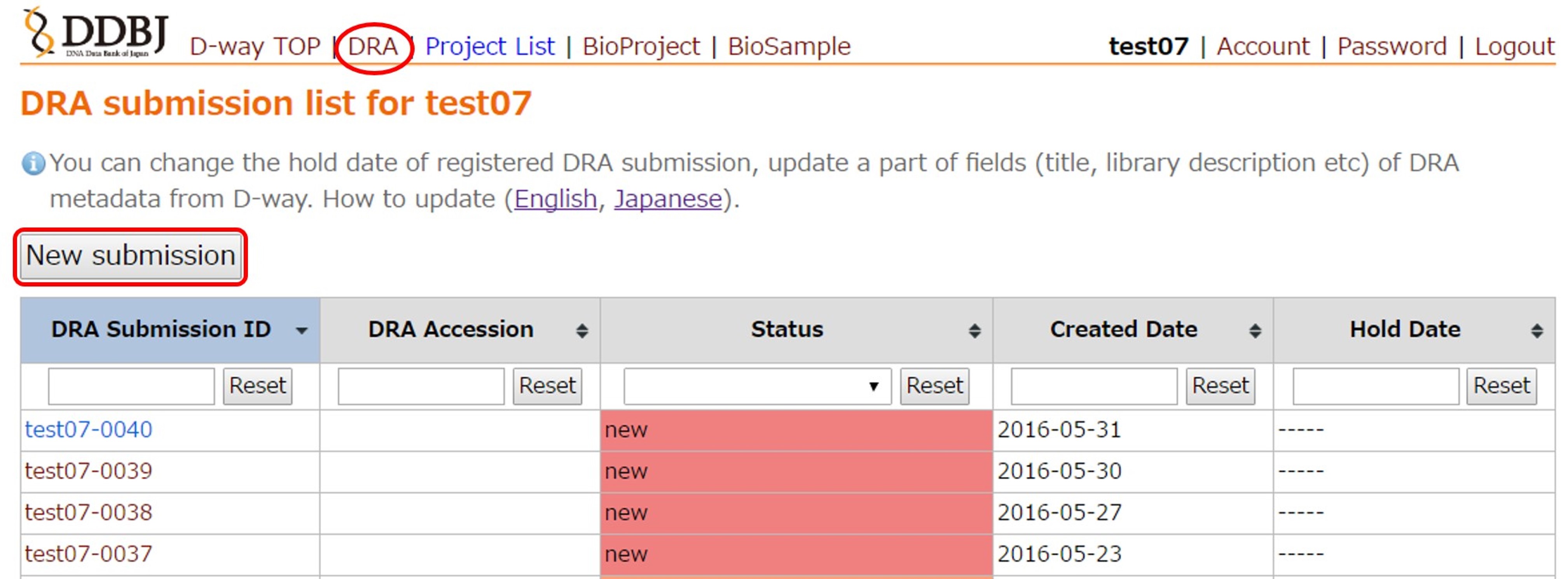
D-way にログインし、上部のメニューから DRA 登録一覧ページへ進みます。
[New submission] で新規登録を作成します。 新規登録に対応するディレクトリ(例 test07-0040/)が受付サーバ (ftp-private.ddbj.nig.ac.jp) に作成されるので、このディレクトリにデータファイルをアップロードします。
- 問い合わせ後、三か月以上回答がない登録はキャンセルします。
- データは Submission 単位で公開されます。データを異なる時期に公開したい場合は Submission を分けてください。
- Submission 辺りの上限は BioSample:1,000、DRA:2,000 (Run 数)、GEA:1,000 (Assay 数) になります。これらを超える件数を登録する場合、同じ BioProject を参照する複数の Submission に分割してください。
データファイルのアップロード
データファイルはファイル受付サーバの対応する Submission ディレクトリ(例 test07-0040/)にアップロードします。 アップロード方法は「データのアップロード」をご覧ください。データをアップロードしないとメタデータを作成することはできません。
登録ステータス
ステータスで進捗状況を把握することができます。”Submission Validated” と “Data Error” になった登録が DRA チームで査定されます。
| ステータス | 説明 |
|---|---|
| New | メタデータの登録前 |
| Metadata Submitted | メタデータが登録された |
| Data Validating | データファイルの検証処理中 |
| Data Error | データファイルの検証処理エラー |
| Submission Validated | メタデータとデータファイルの検証処理を通過 |
| Completed | アクセッション番号が発行された |
| Private | 公開用ファイルの作成が完了し、非公開に保たれている |
| Public | 公開 |
| Canceled | キャンセル |
メタデータの登録
メタデータは複数のオブジェクトから構成されています (構成例)。 BioProject と BioSample は別のデータベースに登録されたレコードを参照します。
- Submission (DRA)
- BioProject
- BioSample
- Experiment (DRA)
- Run (DRA)
- Analysis (DRA、登録は任意)
メタデータは「ウェブツールで登録する方法」と「メタデータ登録用エクセル」を使った二通りの方法があります。 オブジェクト数が多い場合はエクセルを使った方法を推奨します。
以下ではウェブツールでの作成方法を説明します。
Submission ID リンク(例 test07-0040)から登録詳細ページへ移動します。
登録詳細ページ中の [Enter/Update metadata] でメタデータ作成ツールを起動します。
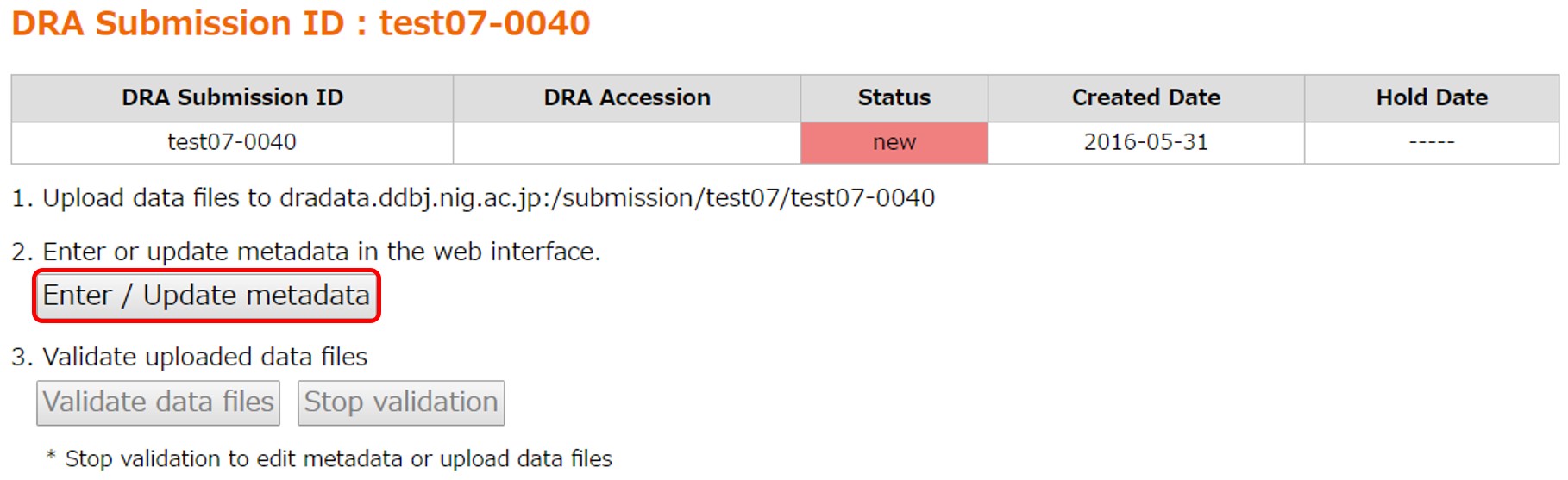
データファイルがアップロードされていない場合、下記のようなメッセージが表示されるので、メタデータを作成する前にデータファイルをアップロードします。

各項目を英語で入力していきます。 必須項目は赤色のアスタリスク記号 (*) で示されています。 画面下部にある [Save] や別のタブへの移動ボタンをクリックすると、都度、入力内容がチェックされます。エラーが表示された場合は内容を修正します。
ウェブツールはタブ区切りテキストファイル (tsv) でのメタデータ入力をサポートしています。 記入例はメタデータ tsv の記入例を参照してください。
Submission
管理情報を記入します。 公開予定、登録者と連絡先メールアドレスを記入します。 Submitting organization (旧 center name) はアカウントの Organiztion の内容が引き写されます。
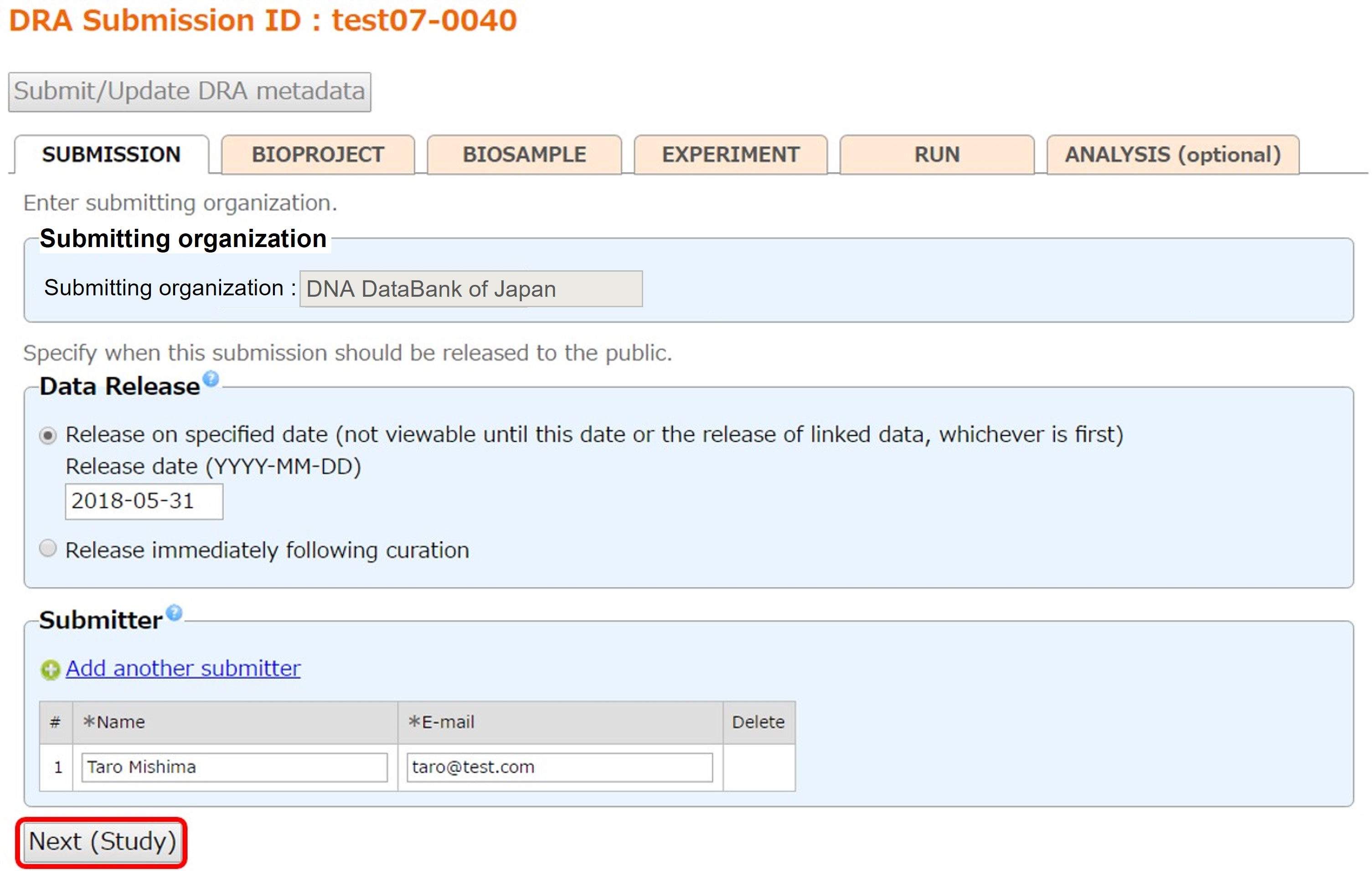
BioProject
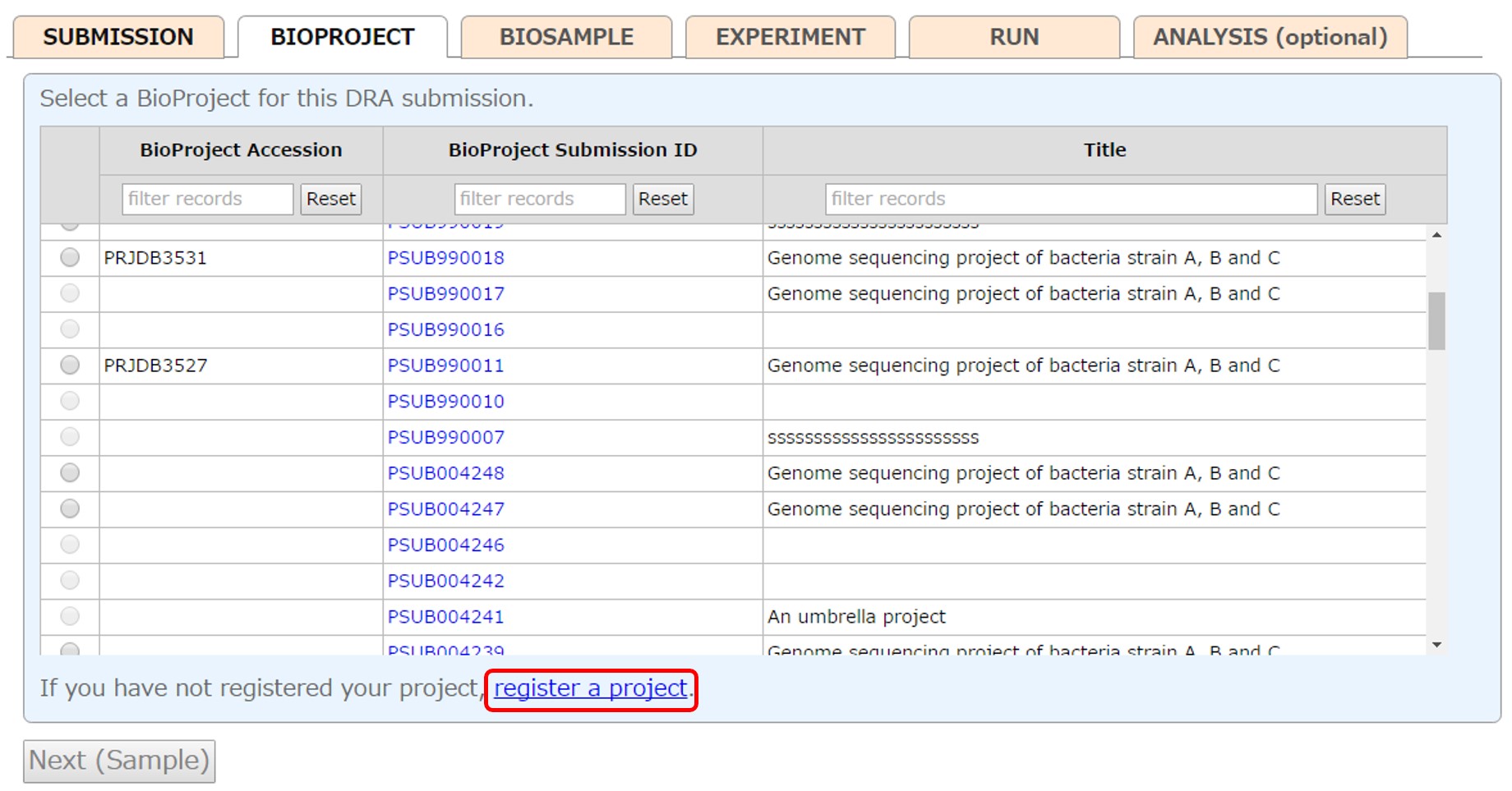
ブラウザーで BioProject が表示されない不具合が生じることがあります。その場合、ブラウザーのプライベートモードや他のブラウザー(Microsoft Edge でうまくいったケースが報告されています)を試してみてください。
アカウントで登録した BioProject から該当するものを1つ選びます。 未登録の場合、[register a project] から新たにプロジェクトを登録します。 自身のアカウント以外で登録された BioProject を参照したい場合は外部参照を申請します。
BioProject の登録方法は「プロジェクトの登録」を参照してください。 登録者と公開予定は DRA Submission で入力した内容が引き写されます。BioProject の登録後、登録したプロジェクトが選択された状態になります。
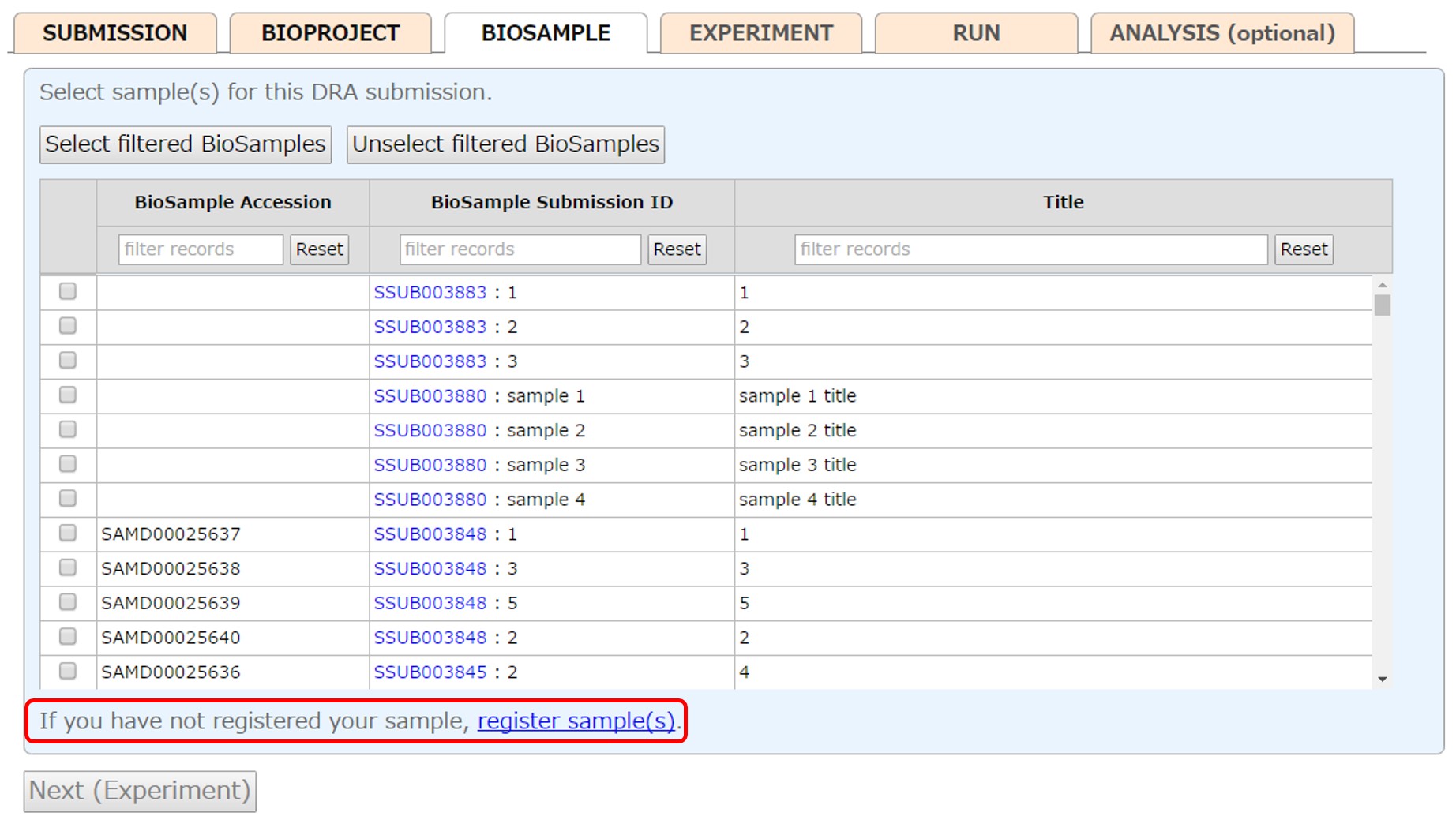
BioSample
ブラウザーで BioSample が表示されない不具合が生じることがあります。その場合、ブラウザーのプライベートモードや他のブラウザー(Microsoft Edge でうまくいったケースが報告されています)を試してみてください。
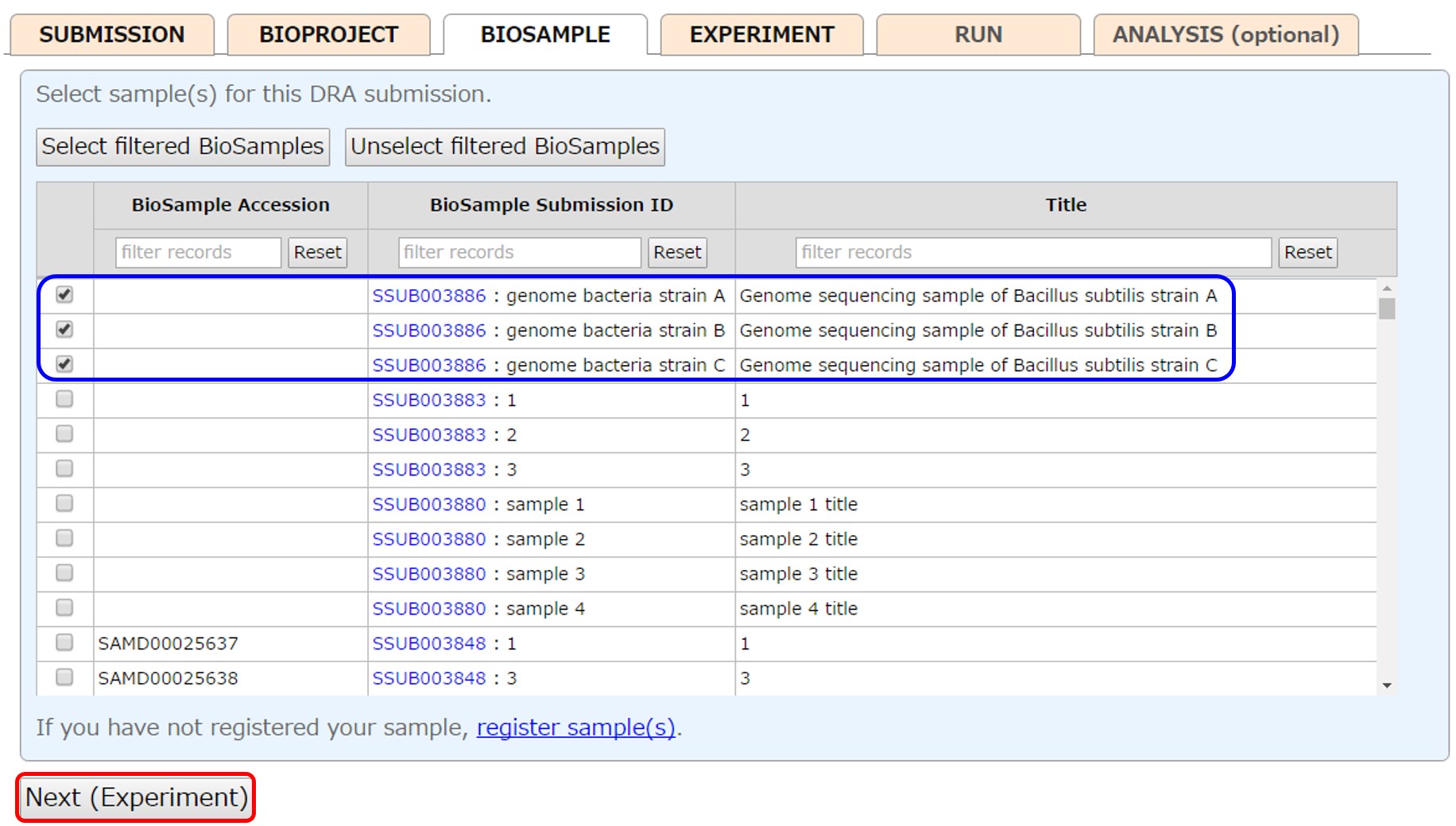
アカウントで登録した BioSample から該当するものを選びます (一般的に DRA ではサンプルは複数になります)。 チェックボックスを選択し、続いて Shift キーを押しながら次のボックスをクリックすると、サンプルが範囲選択されます。 また、カラム上部のボックスにテキストを入力しフィルターされた全てのサンプルを [Select filtered BioSamples] で選択することができます。 未登録の場合、[register sample(s)] から新たにサンプルを登録します。 自身のアカウント以外で登録された BioSample を参照したい場合は外部参照を申請します。
BioSample の登録方法は「サンプルの登録」を参照してください。 登録者と公開予定は DRA Submission で入力した内容が引き写されます。BioSample の登録後、登録したサンプルが選択された状態になります。
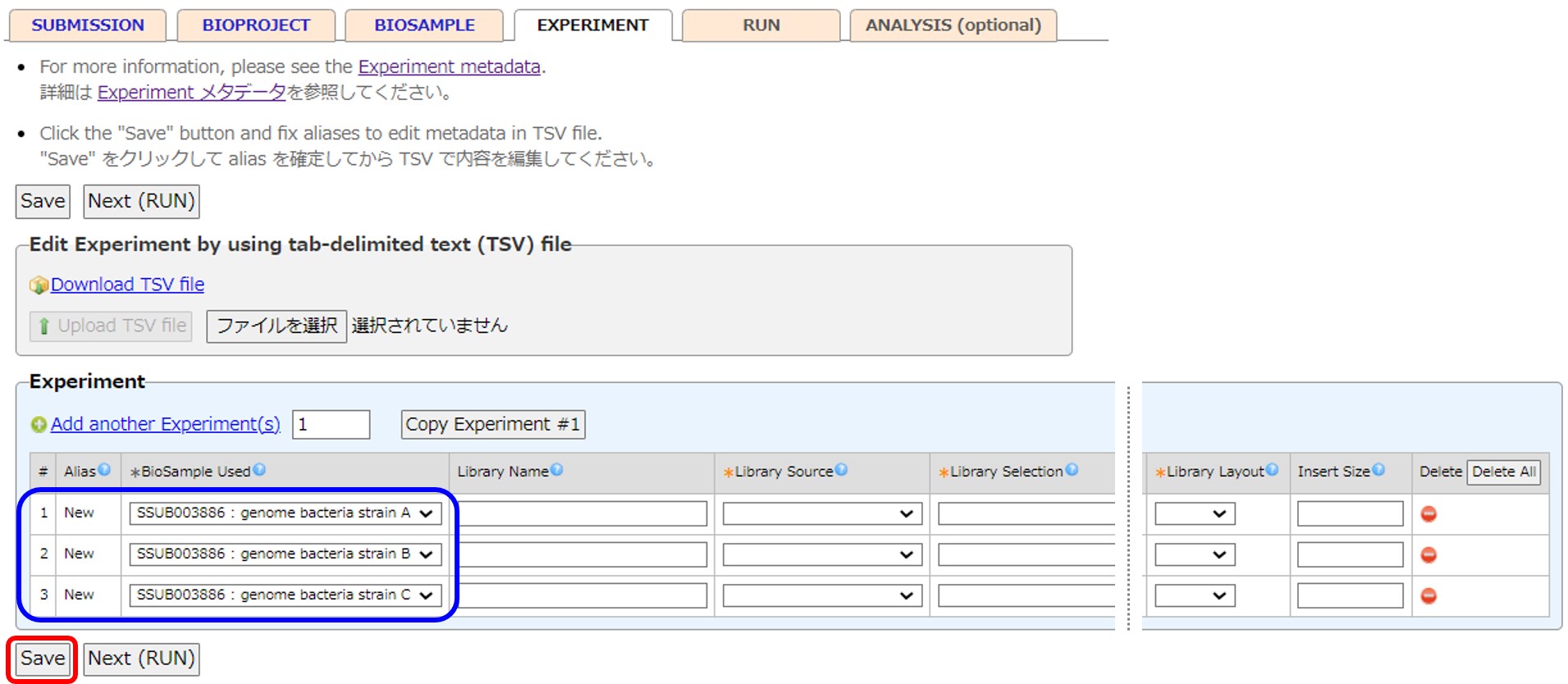
Experiment
初期状態では選択された BioSample と同数の Experiment と Run が自動生成され、それぞれの BioSample-Experiment-Run がリンクされた状態になります。 Experiment と Run の自動生成は Experiment タブの初回表示時にのみ実施され、その後は実施されません。
三つの BioSample を選択した場合の Experiment と Run の自動生成。
| BioProject | - BioSample (1) | - Experiment (1) | - Run (1) |
| - BioSample (2) | - Experiment (2) | - Run (2) | |
| - BioSample (3) | - Experiment (3) | - Run (3) |
Experiment は [Add new Experiment(s)] で追加できます。 また、Experiment は [Delete] で削除することができますが、Run から参照されている Experiment は削除できません。
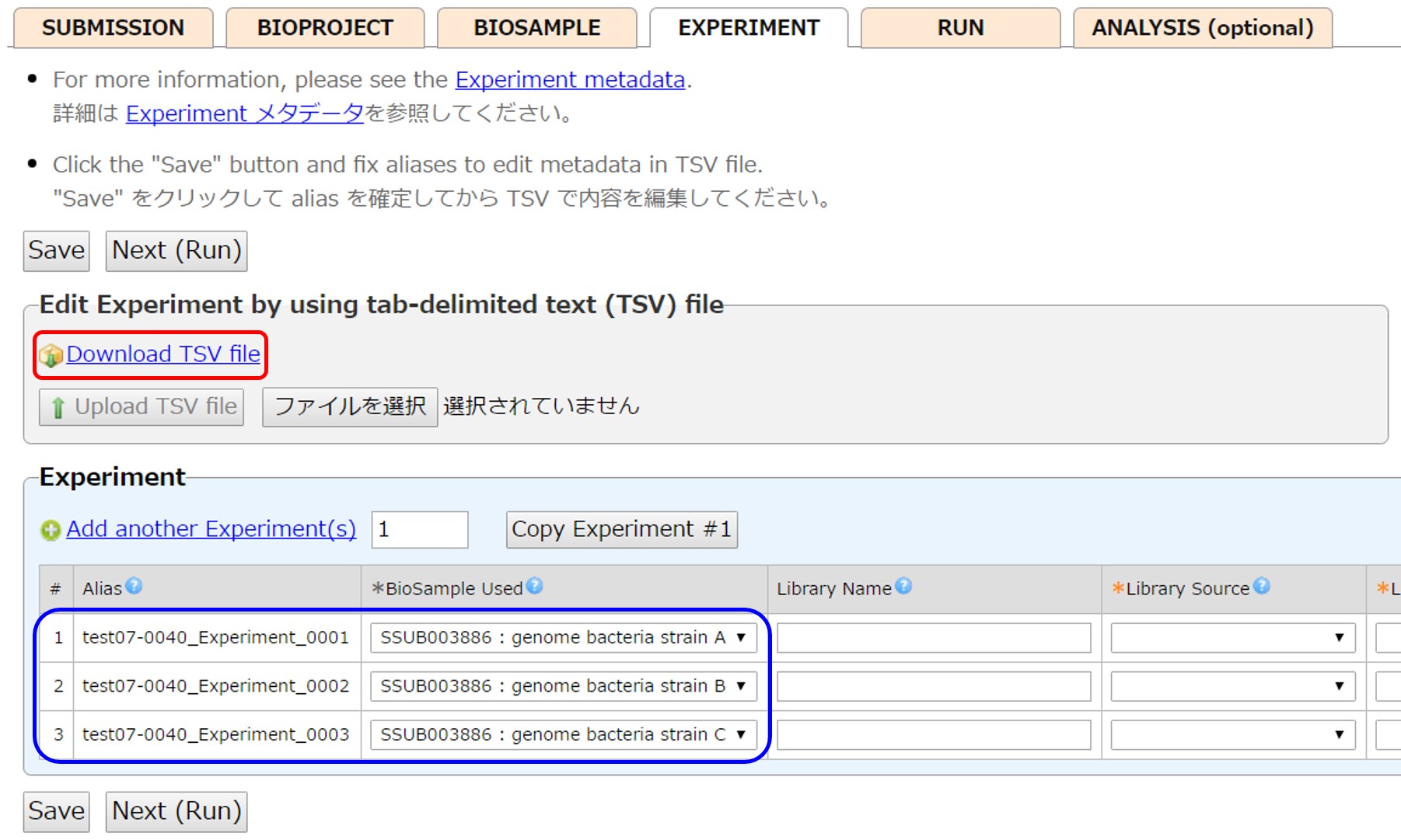
タブ区切りテキストファイルでまとめて Experiment を入力することができます。 まず [Save] で内容を保存し、Alias(アクセッション番号が発行されるまでのオブジェクトの仮の名称。例 test07-0040_Experiment_0001~0003)を確定します。 続いて [Download TSV file] で内容をタブ区切りテキストファイルとしてダウンロードします。
エクセルなどの表計算ソフトでメタデータをまとめて作成します。
“Title” は空の場合、自動的に “[Sequencing Instrument Model] [paired end] sequencing of [BioSample accession]” というタイトル(例 “Illumina HiSeq 2000 paired end sequencing of SAMD00025741”)が構築されます。 内容を簡潔に表すタイトルを “Title” 欄に記載することを推奨します。 “BioSample Used” では BioSample アクセッション番号 (例 SAMD00000001)、もしくは、”SSUB Submission ID” : “Sample name” (例 SSUB003746 : Genome bacteria strain A) の形式で参照する BioSample を指定します。”:” の両側の空白は無視されます。

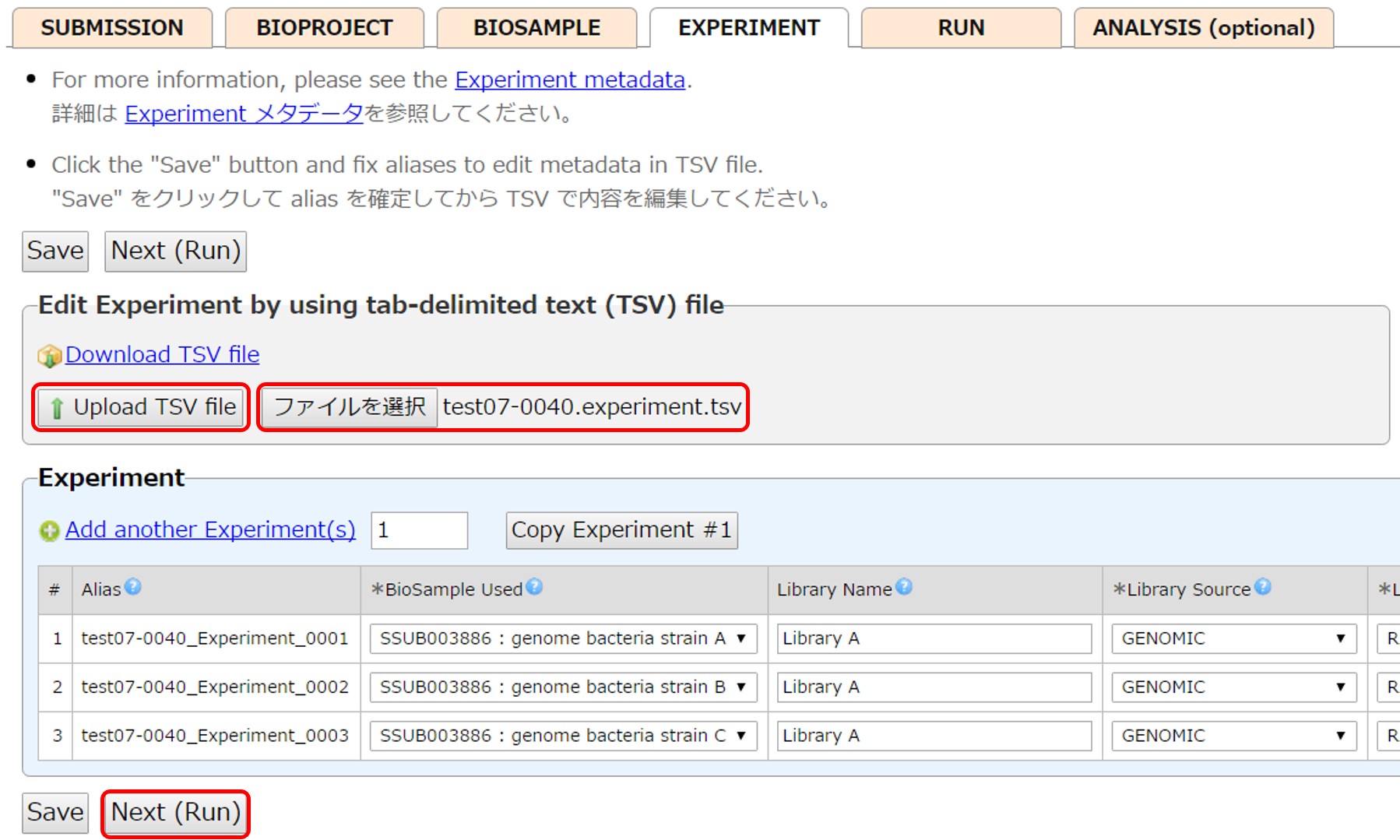
入力内容をタブ区切りテキストファイルとして保存し、選択したうえで [Upload TSV file] で読み込ませます。
Run
初期状態では、選択された Experiment と同数の Run が作成され、それぞれの Experiment を参照しています。下の例では3つの Run が作成され、それぞれが作成された Experiment を参照しています。
[Add another Run(s)] で Run の追加、右端の [Delete] で Run を削除します。ファイルが紐づいている Run は削除することができません。
Run は [Add another Run(s)] で追加できます。 また、Run は [Delete] で削除することができますが、ファイルがリンクしている Run は削除することができません。
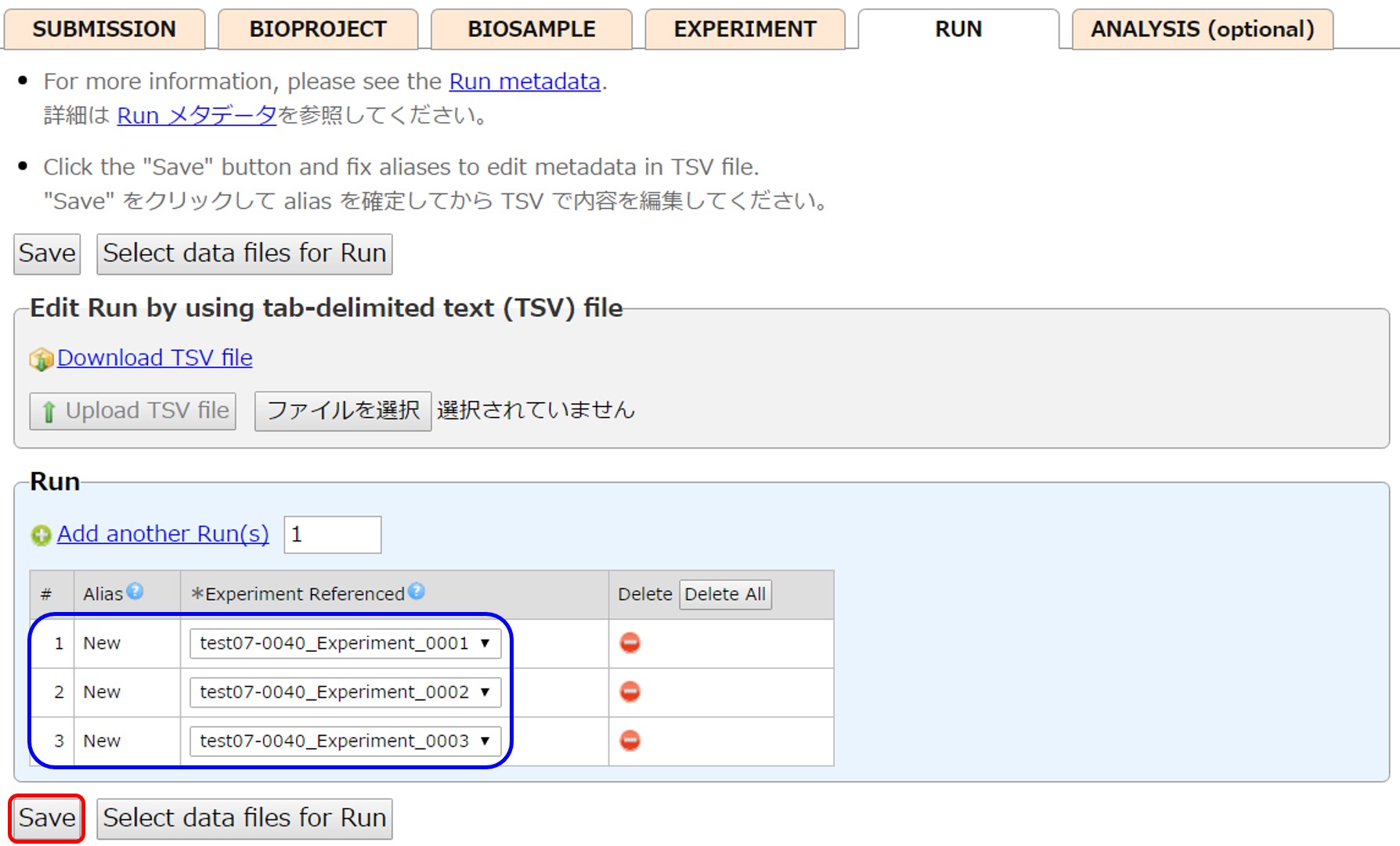
[Save] で Run の Alias を確定すると、内容をタブ区切りテキストファイルで編集できるようになります。 [Select data files for Run] で Run とアップロードしたデータファイルをリンクする画面に移動します。
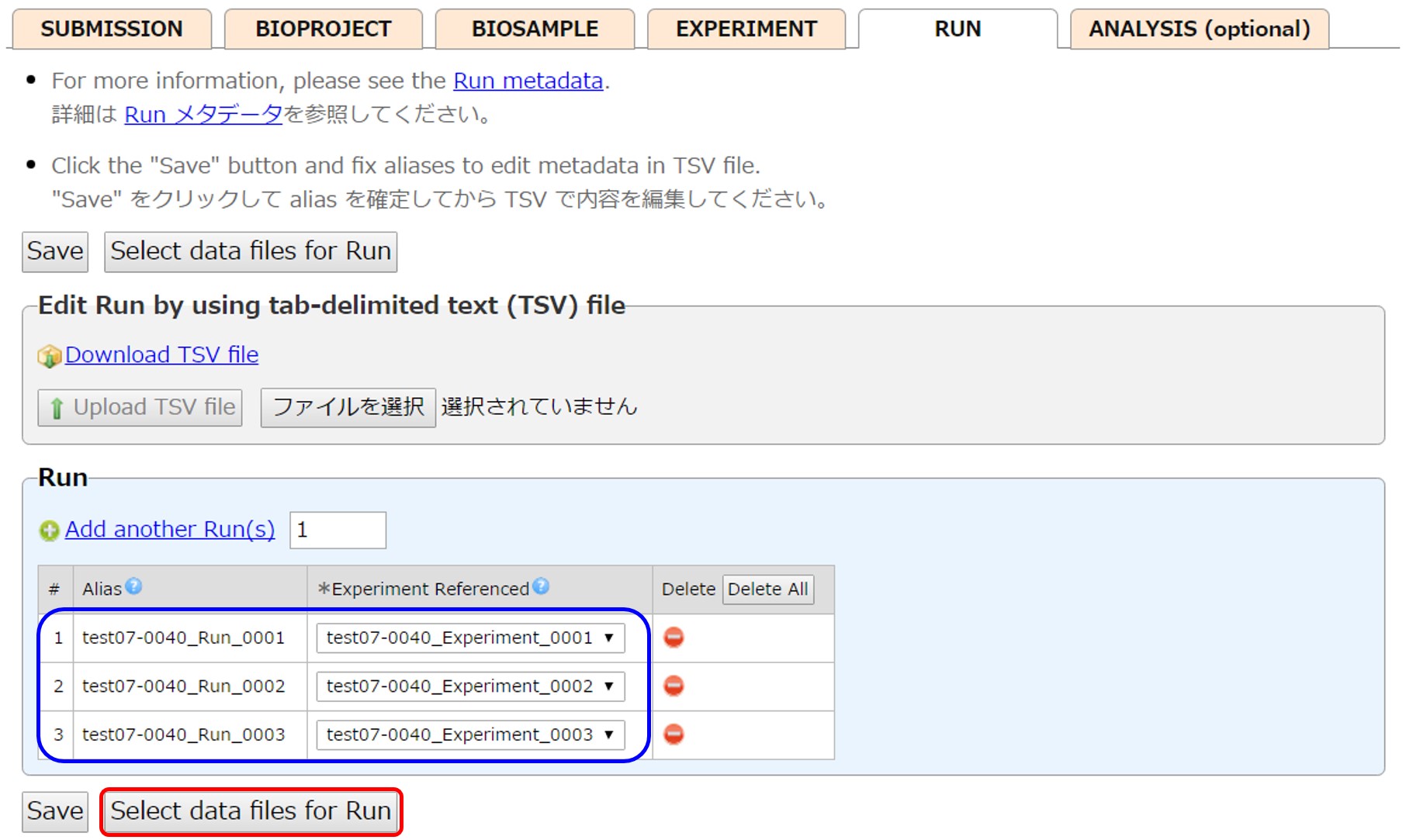
ディレクトリにアップロードされているファイル一覧が表示されます。ファイルが属すべき Run の Alias を “Run/Analysis contains files” で選択します。
続いて File type と MD5 Checksum を入力します。 タブ区切りテキストファイルでまとめて内容を入力する場合、データファイルを区別するため Run に登録するファイルの左端の Run/Analysis に “Run” と入力します。
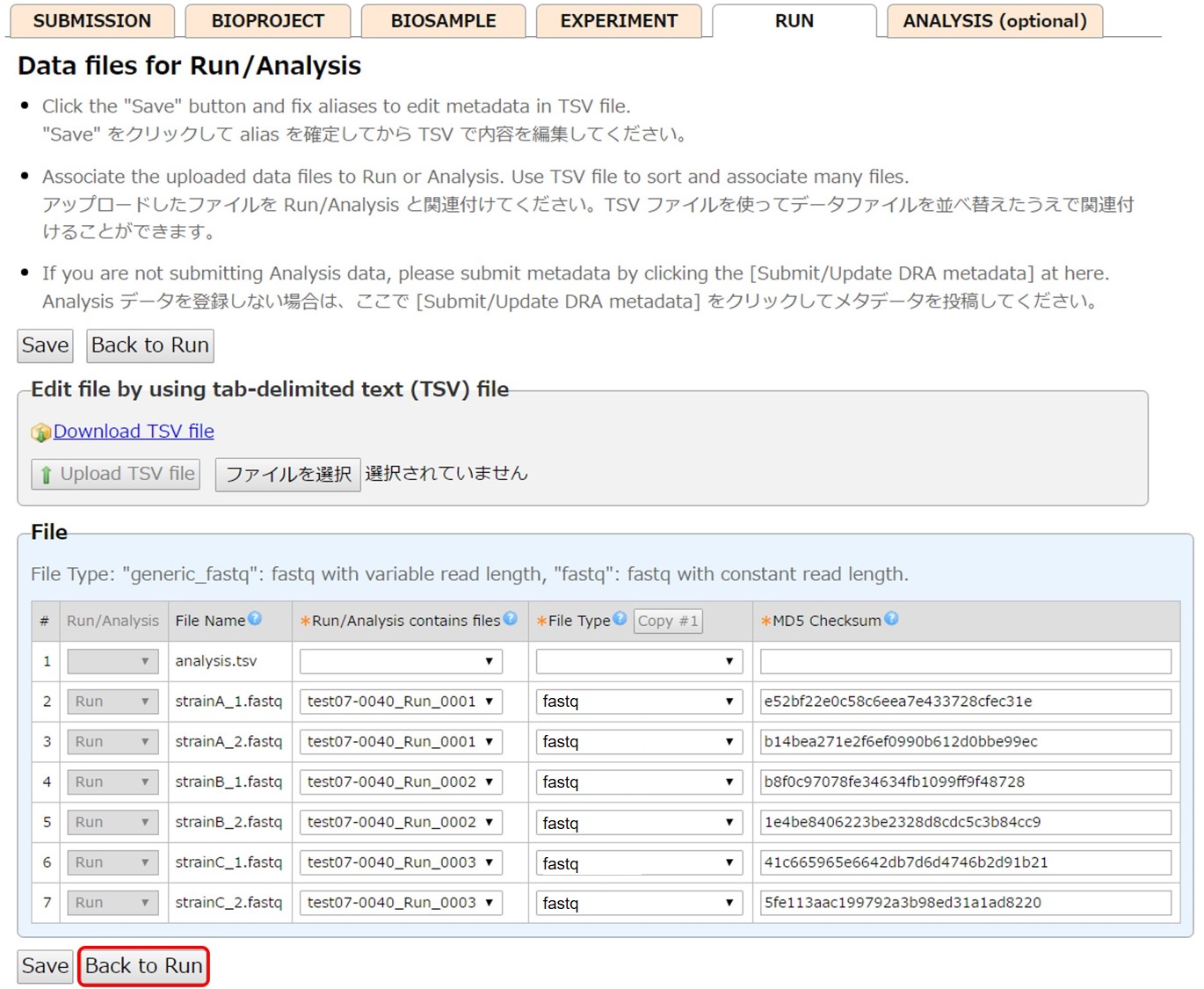
Analysis (任意) が不要な場合は [Submit/Update DRA metadata] でメタデータを登録します。

メタデータの登録後、データファイルの検証処理を開始します。”Validate uploaded data files to finish this submission” をクリックします。

Analysis (任意)
DRA Run に関連するデータで登録先データベースがないデータを Analysis に登録することができます。Analysis は NCBI と EBI で共有していません。 登録先データベースは登録ナビゲーションとデータベースと登録窓口一覧で確認してください。
Analysis を作成後、それぞれの Analysis について内容を記入します。不要な Analysis は [Delete] ボタンで削除します。
[Select data files for Analysis] からファイルと Analysis を結び付ける画面に移動します。
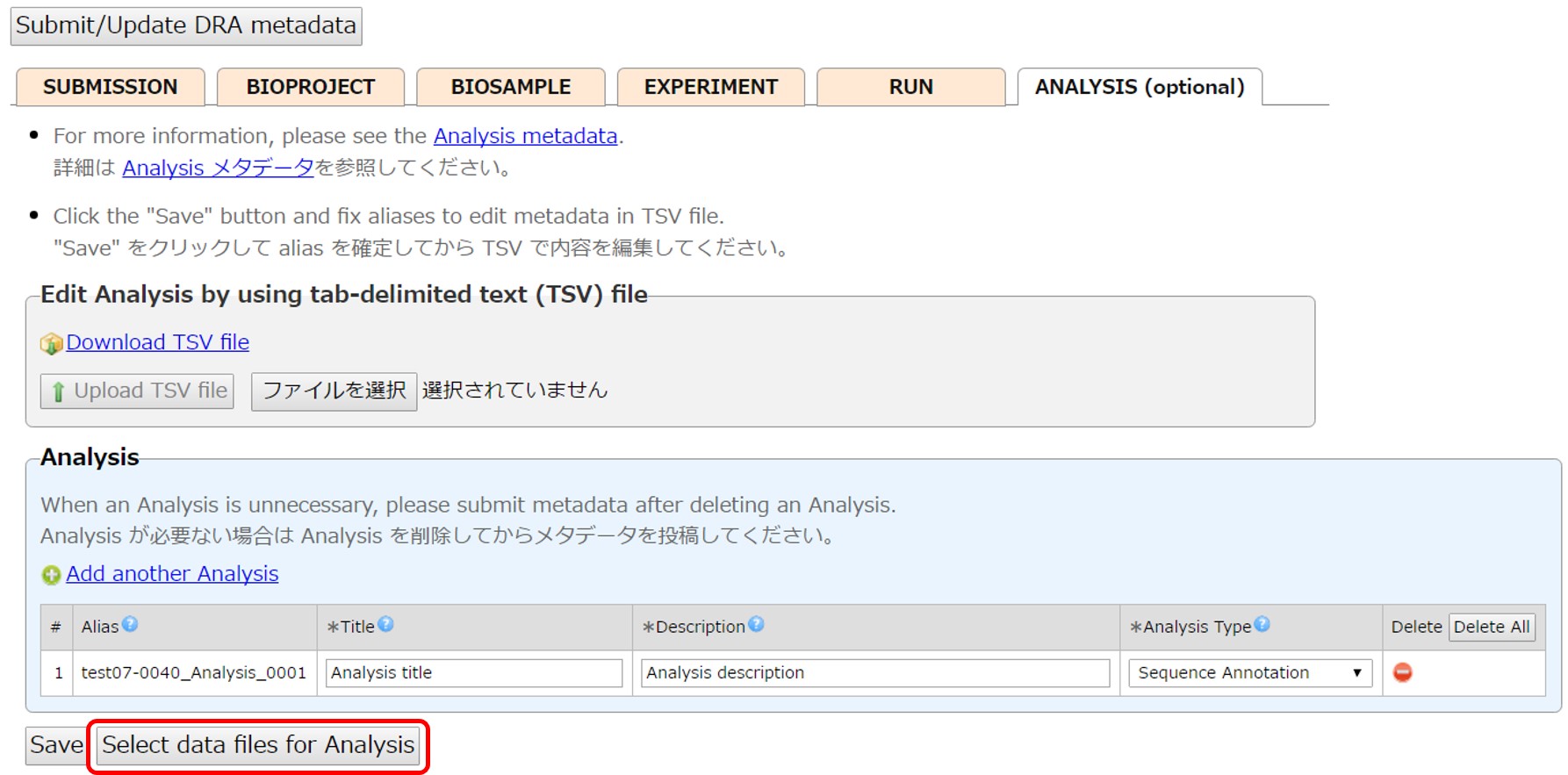
データファイルの属性を入力し、Analysis とリンクさせます。 タブ区切りテキストファイルでまとめて入力する場合、Analysis に登録するファイルを区別するため、左端の Run/Analysis に “Analysis” と入力します。
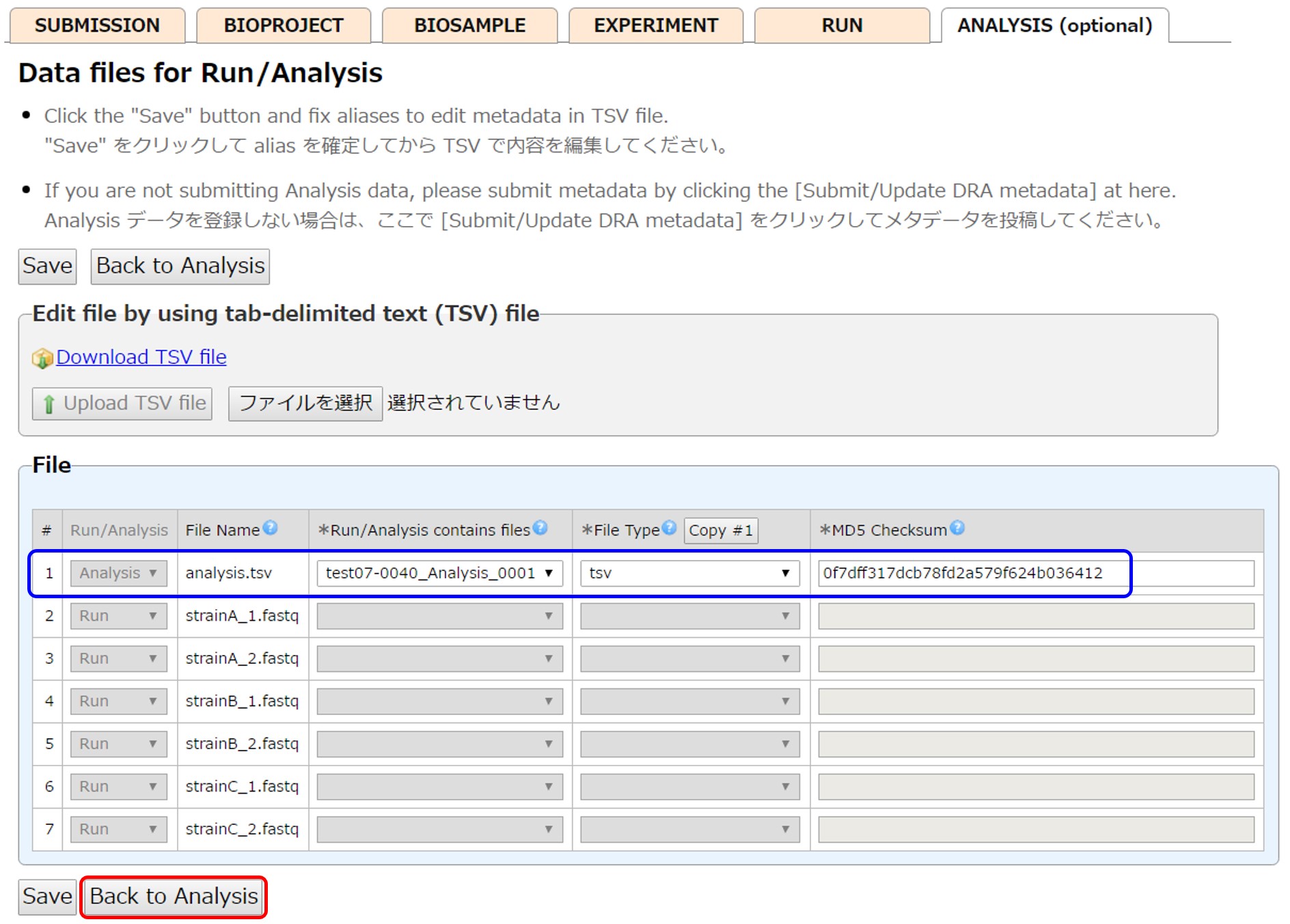
[Enter/Update metadata] で内容を登録し、データファイルの検証処理に進みます。 Analysis に登録されたデータファイルは MD5 チェックサム値の一致チェックのみ実施されます。
エクセルを使った登録方法
ウェブツールと比べて画面遷移が無く、一回の XML ファイルアップロードで登録を済ませることができます。 メタデータ登録用エクセルを記入する前に以下を済ませておきます。
メタデータ登録用エクセルをダウンロードし、内容を記入します。
続いて、以下の手順でエクセルからコマンドラインで XML を生成し、XML を登録します。
手順が分からない方は対象の DRA Submission ID を添え、メール添付でエクセルファイルを DRA 担当者に送付してください。担当者が XML を生成し、登録します。内容を確認後、問題がなければデータファイルの検証処理に進んでください。
GitHub に記載されている手順に従い、エクセルからコマンドラインツールで Submission、Experiment、Run XML を生成します。
ウェブツールやエクセルで入力できない technical read 等の XML 要素を追加する場合、メタデータ XML の例を参照してください。
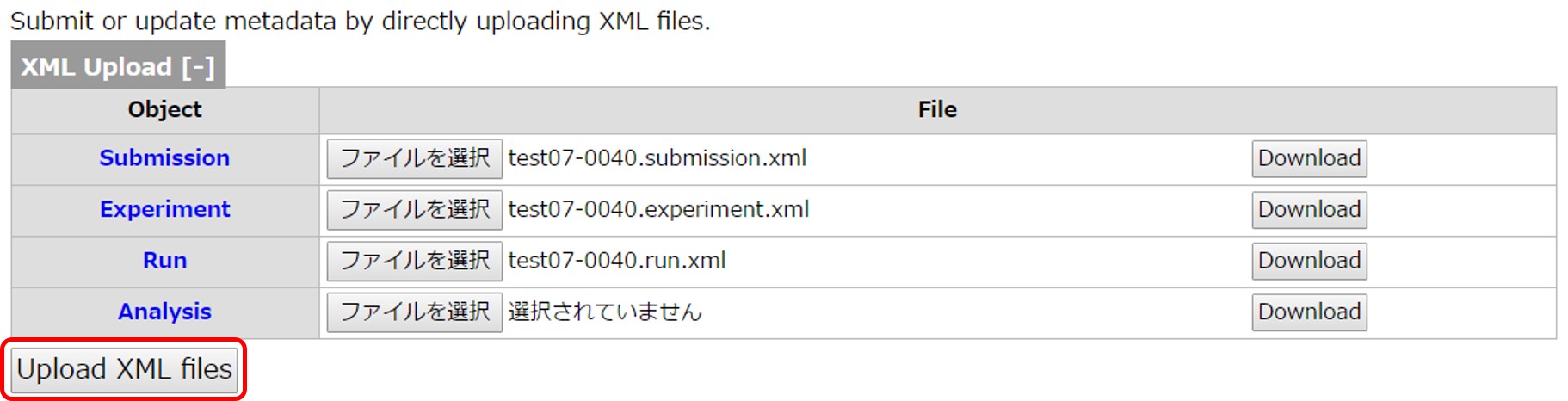
D-way にログインし、対象 Submission のページに移動します。 以下は DRA Submission “test07-0040” に Submission/Experiment/Run XML をアップロードする例です。
検証処理
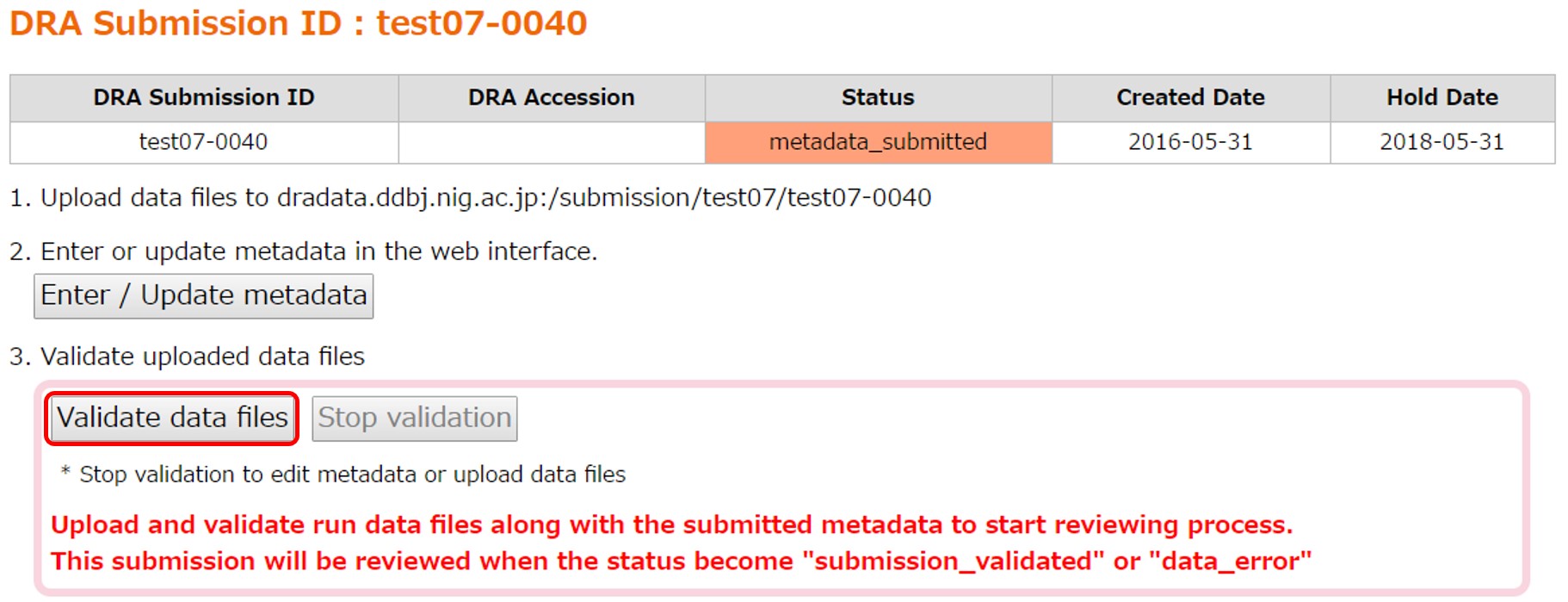
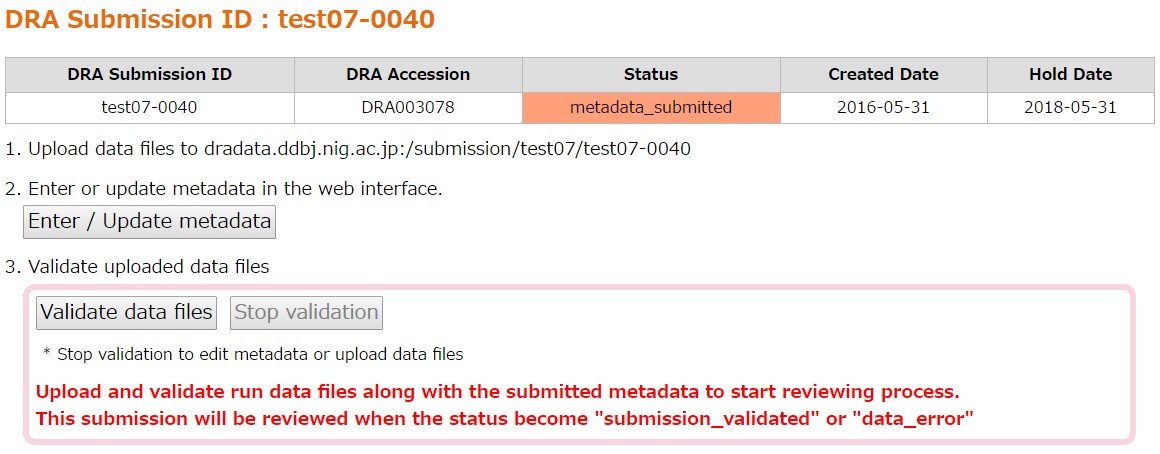
データファイルのチェックサム値とファイルの中身がチェックされます。 “Data Files” に「メタデータに記載されているファイル名と md5 値」及び「受付サーバにアップロードされたファイル名と md5 値」が表示されます。 メタデータに記載されている全てのファイルがアップロードされている場合、[Validate data files] が押下できる状態になります。アップロードされているファイルのうちメタデータに記載されていないものは無視されます。
画面中の [Validate data files] でデータファイルの検証処理を開始します。
MD5 Check
メタデータに記載されている MD5 値と、実際のそれとが一致するかチェックされます。一致しなかった場合はエラーが表示されます。 手許のファイルの MD5 値がメタデータ中のそれと一致している場合、転送過程でファイルが破損した可能性があるため、ファイルを再度アップロードします。メタデータ中の MD5 値が間違っている場合、[Enter/Update metadata] からメタデータ中の MD5 値を修正します。
Data Check
ファイルの中身がチェックされます。 問題が無ければ登録のステータスが “Submission Validated” になり、検証されたファイルが別ディレクトリに移されます。 登録のステータスが “Submission Validated” になると DRA スタッフが査定を始めます。DRA スタッフから指示があるまで D-way を操作せずにお待ちください。
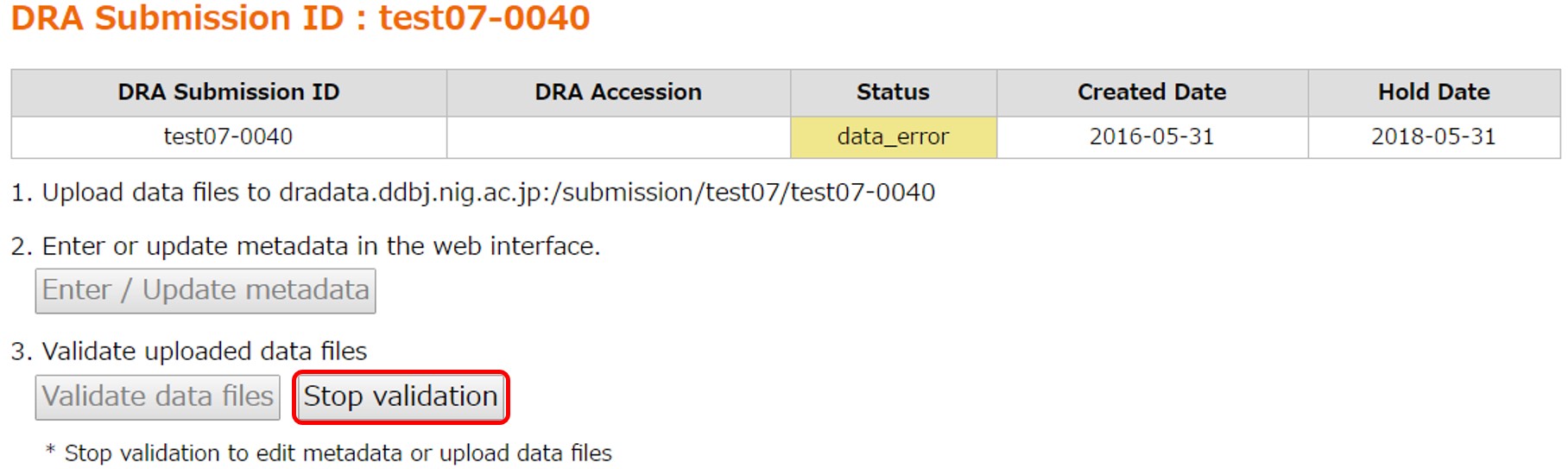
Data Error
検証処理のいずれかのステップでエラーになると、ステータスが “Data Error” になります。 エラーへの対処方法は「FAQ: データファイルの validation エラーへの対処方法は?」を参照してください。 [Stop validation] で検証処理を停止するとステータスが “Metadata Submitted” に戻るので、メタデータの修正やファイルの再アップロードを行い、再度 [Validate data files] をクリックして検証処理を実施します。
アクセッション番号
メタデータとデータファイルに問題がなければ、メタデータオブジェクトにアクセッション番号が発行され、登録者にメールで通知されます。番号は登録詳細ページでも確認することができます。
- Experiment (プレフィックス DRX)
- Run (プレフィックス DRR)
- Analysis (プレフィックス DRZ)
アクセッション番号の引用については「FAQ: 投稿論文ではどのアクセッション番号を引用するべきでしょうか?」をご覧ください。
データ公開
データの処理とステータスについては DDBJ と SRA におけるデータ処理をご覧ください。
データは公開原則に従って公開されます。
参照関係にあるデータとの連動公開については、「FAQ: BioProject/BioSample/塩基配列データの連動公開の仕組みは?」を参照してください。
公開されるとデータが ftp に公開され、数日以内に DDBJ Search でインデックスされ、NCBI SRA と EBI SRA にミラーリングされます。
登録済みファイルの提供
登録済みデータを確認できるようにするため、登録が完了した Run の fastq/sra ファイルは受付サーバ (ftp-private.ddbj.nig.ac.jp) の登録者領域にコピーされます。ディスク容量圧迫を防ぐため、コピーされたファイルは一ヶ月後に削除されます。
- /report/dra/(DRA submission accession)/fastq/
- /report/dra/(DRA submission accession)/sra/
例 /report/dra/DRA000001/fastq/DRR000001.fastq.bz2
更新
公開予定日延長、メタデータの更新やデータの追加・削除については DRA の更新をご覧ください。