Sequence Read Archive
メタデータ
オブジェクト
メタデータにはシークエンスデータがどのようにして得られたのかが記載されています。 メタデータは複数のオブジェクトから構成され、各オブジェクトは XML スキーマで定義され、相互に関連付けられています。メタデータの例
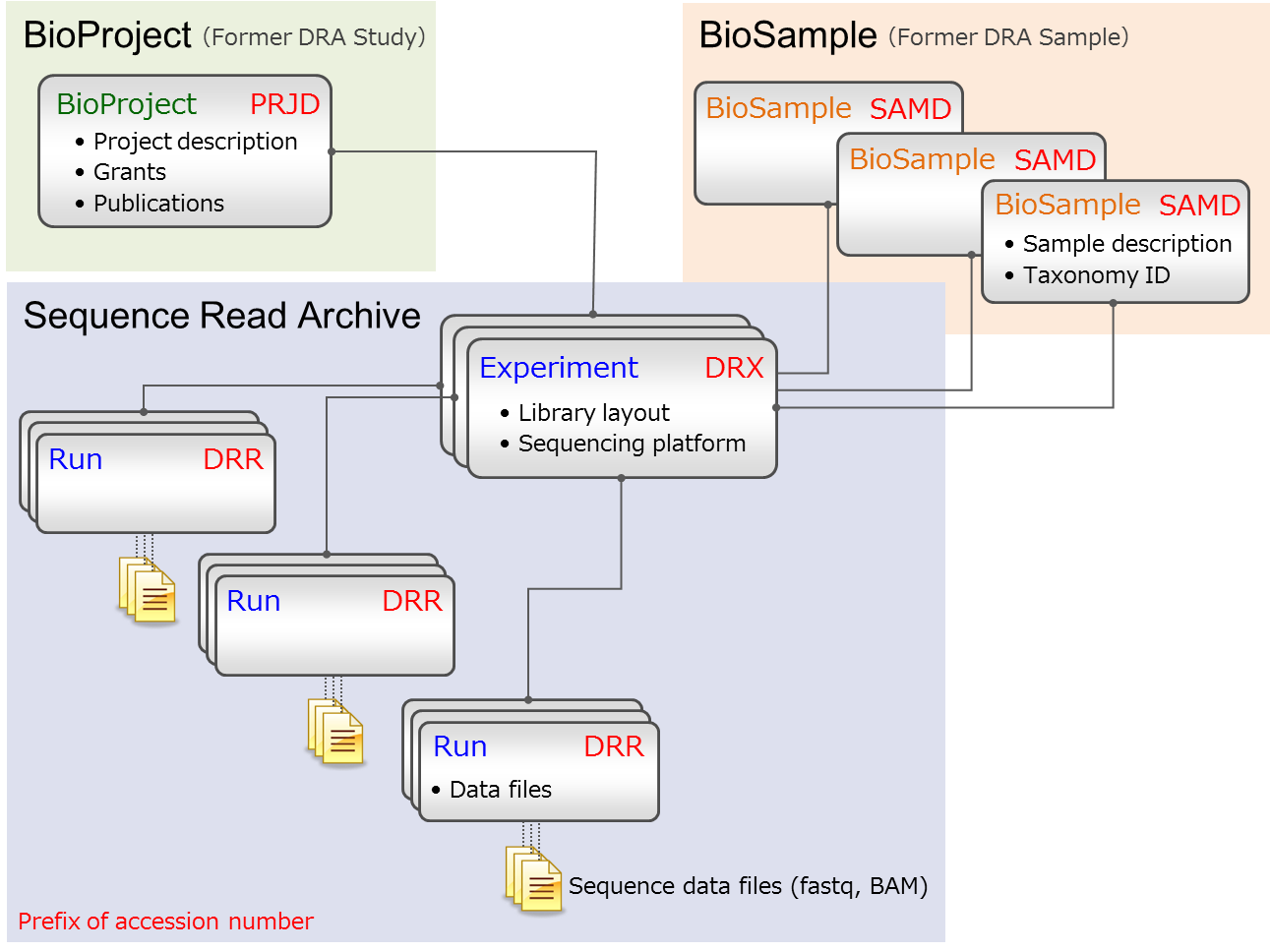
- Submission
- 公開予定や登録者といった管理情報を記載し、同時に登録する DRA オブジェクトをまとめる。
- BioProject
- 研究プロジェクト。外部の BioProject データベース。
- BioSample
- シークエンスデータが得られた生物学的なサンプル。外部の BioSample データベース。
- Experiment
- サンプルから構築されたライブラリーとシークエンス機種に関する情報を記載。Experiment は1つの BioProject と1つの BioSample を参照します。 複数の Experiment は1つの BioSample を参照することができますが、1つの Experiment が複数の BioSample を参照することはできません。
- Run
- シークエンス用ライブラリー (Experiment) に由来するデータファイルをまとめます。Experiment を介してデータファイルは特定のサンプルにリンクされます。 Run に含まれる全てのファイルは1つの sra/fastq ファイルに変換され、Run のアクセッション番号がファイル名になります。 ペアードデータファイルは同じ Run に含め、リードがペアとして処理されるようにします。
- Analysis
- Run データに関連するデータで登録先がないデータを登録します。Analysis は DDBJ/EBI/NCBI で交換していません。
メタデータの項目
必須* 条件によって必須*
Submission
Submitting organization
アカウントの Organization が自動的に引き写されます。 2023年12月20日に center name は廃止され、組織名の略号管理はなくなりました。
Hold Until
公開方法を指定します。
- Hold Until*
- 公開予定日を設定します。最長で4年後まで設定でき、延長することができます。
- Immediate Release*
- 即日公開。登録処理が完了次第、データが公開されます。
Submitter
登録者の名前とメールアドレス。責任者 (principal investigator) を含めてください。登録に関する連絡は記入された全てのアドレスに対して行われます。 登録者情報は公開されません。
DRA 登録に関するメールは Submission に記載されたメールアドレスに対して送信されます。
DDBJ アカウントに登録されているメールアドレスを変更した場合、登録に関するメールが送信されるように Submission のアドレスも変更する必要があります。
BioProject
- BioProject ID*
- BioProject に登録済みのプロジェクトから該当するものを1つ選択、もしくは、新たに登録します。BioProject の登録方法は BioProject の登録を参照してください。
BioSample
- BioSample ID*
- BioSample に登録済みのサンプルから該当するものを選択、もしくは、新たに登録します。BioSample の登録方法は BioSample の登録を参照してください。
Experiment
- Alias
- 自動的に Experiment に付けられる名前。アクセッション番号のないオブジェクトは Alias で参照されます。
- BioSample Used*
- Experiment が参照している BioSample を選択します。
- Title*
- 検索結果で表示される Experiment のタイトル。 自動的に “[Sequencing Instrument Model] [paired end] sequencing of [BioSample ID]” というタイトル(例 “Illumina HiSeq 2000 paired end sequencing of SAMD00025741”)が構築されます。 独自のタイトルを入力する場合は、Experiment の内容をタブ区切りテキストファイルとしてダウンロードし、Title カラムにタイトルを入力し、アップロードします。
- Library Name
- ライブラリーの名前。
- Library Source*
- ライブラリー構築に用いた試料。
| Library Source | Description |
|---|---|
| GENOMIC | Genomic DNA (includes PCR products from genomic DNA). |
| GENOMIC SINGLE CELL | Genomic DNA from single cell. |
| TRANSCRIPTOMIC | Transcription products or non genomic DNA (EST, cDNA, RT-PCR, screened libraries). |
| TRANSCRIPTOMIC SINGLE CELL | Transcription products or non genomic DNA from single cell. |
| METAGENOMIC | Mixed material from metagenome. |
| METATRANSCRIPTOMIC | Transcription products from community targets. |
| SYNTHETIC | Synthetic DNA. |
| VIRAL RNA | Viral RNA. |
| OTHER | Other, unspecified, or unknown library source material. |
- Library Selection*
- シークエンスに用いたライブラリを構築するためのサンプルの選別や濃縮方法。
| Library Selection | Description |
|---|---|
| RANDOM | Random shearing only. |
| PCR | Source material was selected by designed primers. |
| RANDOM PCR | Source material was selected by randomly generated primers. |
| RT-PCR | Source material was selected by reverse transcription PCR. |
| HMPR | Hypo-methylated partial restriction digest. |
| MF | Methyl Filtrated. |
| repeat fractionation | Selection for less repetitive (and more gene rich) sequence through Cot filtration (CF) or other fractionation techniques based on DNA kinetics. |
| size fractionation | Physical selection of size appropriate targets. |
| MSLL | Methylation Spanning Linking Library. |
| cDNA | complementary DNA. |
| cDNA_randomPriming | |
| cDNA_oligo_dT | |
| PolyA | PolyA selection or enrichment for messenger RNA (mRNA); should replace cDNA enumeration. |
| Oligo-dT | enrichment of messenger RNA (mRNA) by hybridization to Oligo-dT. |
| Inverse rRNA | depletion of ribosomal RNA by oligo hybridization. |
| ChIP | Chromatin immunoprecipitation. |
| MNase | Micrococcal Nuclease (MNase) digestion. |
| DNAse | Deoxyribonuclease (DNase) digestion. |
| Hybrid Selection | Selection by hybridization in array or solution. |
| Reduced Representation | Reproducible genomic subsets, often generated by restriction fragment size selection, containing a manageable number of loci to facilitate re-sampling. |
| Restriction Digest | DNA fractionation using restriction enzymes. |
| 5-methylcytidine antibody | Selection of methylated DNA fragments using an antibody raised against 5-methylcytosine or 5-methylcytidine (m5C)MBD2 protein methyl-CpG binding domain : Enrichment by methyl-CpG binding domain. |
| MBD2 protein methyl-CpG binding domain | MBD2 protein methyl-CpG binding domain. |
| CAGE | Cap-analysis gene expression. |
| RACE | Rapid Amplification of cDNA Ends. |
| MDA | multiple displacement amplification. |
| padlock probes capture method | Padlock Probes capture strategy to be used in conjuction with Bisulfite-Seq. |
| other | Other library enrichment, screening, or selection process. |
| unspecified | Library enrichment, screening, or selection is not specified. |
- Library Strategy*
- ライブラリーの構築手法。
| Library Strategy | Description |
|---|---|
| WGS | Whole genome shotgun. |
| WGA | Whole genome amplification. |
| WXS | Random sequencing of exonic regions selected from the genome. |
| RNA-Seq | Random sequencing of whole transcriptome. |
| miRNA-Seq | Micro RNA and other small non-coding RNA sequencing. |
| ncRNA-Seq | Capture of other non-coding RNA types, including post-translation modification types such as snRNA (small nuclear RNA) or snoRNA (small nucleolar RNA), or expression regulation types such as siRNA (small interfering RNA) or piRNA/piwi/RNA (piwi-interacting RNA). |
| ssRNA-seq | strand-specific RNA sequencing |
| WCS | Whole chromosome (or other replicon) shotgun. |
| CLONE | Genomic clone based (hierarchical) sequencing. |
| POOLCLONE | Shotgun of pooled clones (usually BACs and Fosmids). |
| AMPLICON | Sequencing of overlapping or distinct PCR or RT-PCR products. |
| CLONEEND | Clone end (5’, 3’, or both) sequencing. |
| FINISHING | Sequencing intended to finish (close) gaps in existing coverage. |
| RAD-Seq | Restriction Site Associated DNA Sequence |
| ChIP-Seq | Direct sequencing of chromatin immunoprecipitates. |
| MNase-Seq | Direct sequencing following MNase digestion. |
| DNase-Hypersensitivity | Sequencing of hypersensitive sites, or segments of open chromatin that are more readily cleaved by DNaseI. |
| Bisulfite-Seq | Sequencing following treatment of DNA with bisulfite to convert cytosine residues to uracil depending on methylation status. |
| EST | Single pass sequencing of cDNA templates. |
| FL-cDNA | Full-length sequencing of cDNA templates. |
| CTS | Concatenated Tag Sequencing. |
| MRE-Seq | Methylation-Sensitive Restriction Enzyme Sequencing strategy. |
| MeDIP-Seq | Methylated DNA Immunoprecipitation Sequencing strategy. |
| MBD-Seq | Direct sequencing of methylated fractions sequencing strategy. |
| Tn-Seq | Gene fitness determination through transposon seeding. |
| FAIRE-seq | Formaldehyde Assisted Isolation of Regulatory Elements |
| SELEX | Systematic Evolution of Ligands by EXponential enrichment |
| NOMe-Seq | Nucleosome Occupancy and Methylome sequencing. |
| RIP-Seq | Direct sequencing of RNA immunoprecipitates (includes CLIP-Seq, HITS-CLIP and PAR-CLIP). |
| ChIA-PET | Direct sequencing of proximity-ligated chromatin immunoprecipitates. |
| Hi-C | Chromosome Conformation Capture technique where a biotin-labeled nucleotide is incorporated at the ligation junction, enabling selective purification of chimeric DNA ligation junctions followed by deep sequencing |
| ATAC-seq | Assay for Transposase-Accessible Chromatin (ATAC) strategy is used to study genome-wide chromatin accessibility. alternative method to DNase-seq that uses an engineered Tn5 transposase to cleave DNA and to integrate primer DNA sequences into the cleaved genomic DNA |
| Targeted-Capture | |
| Tethered Chromatin Conformation Capture | |
| Synthetic-Long-Read | binning and barcoding of large DNA fragments to facilitate assembly of the fragment |
| Other | Library strategy not listed. |
- Library Construction Protocol
- DNA の断片化、アダプター配列などのライゲーション (DNA ligation) や濃縮 (DNA enrichment) 方法をフリーテキストで記載します。キットを使用した場合はキットの名前とバージョン (あれば) を含めます (例 Illumina Nextera DNA Library Preparation Kit)。
参考: Alnasir J, Shanahan HP. Investigation into the annotation of protocol sequencing steps in the sequence read archive. Gigascience. 2015 May 9;4:23. doi: 10.1186/s13742-015-0064-7. eCollection 2015. PMID: 25960871 (Open Access)
- Instrument*
- シークエンサの機種を選択します。
| Instrument Model |
|---|
| 454 GS |
| 454 GS 20 |
| 454 GS FLX |
| 454 GS FLX+ |
| 454 GS FLX Titanium |
| 454 GS Junior |
| Illumina Genome Analyzer |
| Illumina Genome Analyzer II |
| Illumina Genome Analyzer IIx |
| Illumina HiSeq 1000 |
| Illumina HiSeq 1500 |
| Illumina HiSeq 2000 |
| Illumina HiSeq 2500 |
| Illumina HiSeq 3000 |
| Illumina HiSeq 4000 |
| HiSeq X Five |
| HiSeq X Ten |
| Illumina HiSeq X |
| Illumina HiScanSQ |
| Illumina NovaSeq 6000 |
| Illumina NovaSeq X |
| Illumina NovaSeq X Plus |
| Illumina MiSeq |
| Illumina MiSeq i100 |
| Illumina MiSeq i100 Plus |
| Illumina MiniSeq |
| Illumina iSeq 100 |
| NextSeq 500 |
| NextSeq 550 |
| NextSeq 1000 |
| NextSeq 2000 |
| Helicos HeliScope |
| AB SOLiD System |
| AB SOLiD System 2.0 |
| AB SOLiD System 3.0 |
| AB SOLiD 3 Plus System |
| AB SOLiD 4 System |
| AB SOLiD 4hq System |
| AB SOLiD PI System |
| AB 5500 Genetic Analyzer |
| AB 5500xl Genetic Analyzer |
| AB 5500xl-W Genetic Analysis System |
| Complete Genomics |
| BGISEQ-50 |
| BGISEQ-500 |
| MGISEQ-2000RS |
| CycloneSEQ |
| DNBSEQ-G400 |
| DNBSEQ-G400 FAST |
| DNBSEQ-T7 |
| DNBSEQ-T1+ |
| DNBSEQ-G50 |
| PacBio RS |
| PacBio RS II |
| Sequel |
| Sequel II |
| Sequel IIe |
| Onso |
| Revio |
| Ion Torrent PGM |
| Ion Torrent Proton |
| Ion Torrent S5 |
| Ion Torrent S5 XL |
| Ion GeneStudio S5 |
| Ion GeneStudio S5 plus |
| Ion GeneStudio S5 prime |
| Ion Torrent Genexus |
| MinION |
| GridION |
| PromethION |
| GENIUS |
| Genapsys Sequencer |
| GS111 |
| Sentosa SQ301 |
| Element AVITI |
| GenoCare 1600 |
| GenoLab M |
| FASTASeq 300 |
| UG 100 |
| Tapestri |
| SURFSeq 5000 |
| SURFSeq Q |
| Saluseq Nimbo |
| Salus Pro |
| Salus EVO |
| G-seq500 |
| G4 |
| AB 3730xL Genetic Analyzer |
| AB 3730 Genetic Analyzer |
| AB 3500xL Genetic Analyzer |
| AB 3500 Genetic Analyzer |
| AB 3130xL Genetic Analyzer |
| AB 3130 Genetic Analyzer |
| AB 310 Genetic Analyzer |
- Library Layout*
- データファイル中のリード構成を選択します。リードの向き (Forward と Reverse) は Instrument から自動判定されます。2022年12月に Spot Type から Library Layout に表示名が変更になりました。
| Spot Type | Description |
|---|---|
| single | Single read |
| paired | Paired reads |
- Insert Size*
- ペアエンドライブラリを構築した際のインサートの平均サイズ。2022年12月に Nominal Length から Insert Size に表示名が変更になりました。
Run
- Alias
- 自動的に Run に付けられる名前。アクセッション番号のないオブジェクトは Alias で参照されます。
- Title*
- Run の短いタイトル。ユニークなタイトルを付けます。 検索結果で表示される Run の短いタイトル。 自動的に “[Sequencing Instrument Model] [paired end] sequencing of [BioSample ID]” というタイトル(例 “Illumina HiSeq 2000 paired end sequencing of SAMD00025741”)が構築されます。 独自のタイトルを入力する場合は、Run の内容をタブ区切りテキストファイルとしてダウンロードし、Title カラムにユニークなテキストを入力し、アップロードします。
- Experiment Referenced*
- Run が属する Experiment を選択します。
Data files for Run
Run に含めるデータファイルを選択します。
- Run/Analysis
- データファイルが Run もしくは Analysis に属しているのかを指定します。ウェブ画面上では入力できず、属している Run もしくは Analysis の alias が選択されると自動的に入力されます。タブ区切りテキストファイルで入力する場合には、Run もしくは Analysis を入力します。
- File Name*
- シークエンスデータファイル名。DRA サーバにアップロードされているファイル名が自動的に入力されます。
- Run/Analysis contains files*
- データファイルが属する Run を選択します。
- File Type*
- シークエンスデータのファイル形式。fastq ファイルの場合、リード長が一定かそうでないかに関わらず全て “fastq” を選択します。
| File Type | Description |
|---|---|
| fastq | fastq files |
| hdf5 | PacBio hdf5 Format file |
| bam | Binary SAM format for use by loaders that combine alignment and sequencing data |
| tab | A tab-delimited table maps “SN in SQ line of BAM header” and “reference fasta file” |
| reference_fasta | Reference sequence file in single fasta format used to construct SRA archive file format. Filename must end with “.fa” |
- MD5 Checksum*
- データファイルの MD5 チェックサム値。MD5 チェックサム値の取得方法
Analysis
- Alias
- 自動的に Analysis に付けられる名前。アクセッション番号のないオブジェクトは Alias で参照されます。
- Title*
- Analysis オブジェクトのタイトル。
- Description*
- Analysis の内容を記述します。
- Analysis Type*
- Analysis の種類を選択します。アライメントデータは Run に登録します。
| Analysis Type | Description |
|---|---|
| De Novo Assembly | A placement of sequences including trace, SRA, GI records into a multiple alignment from which a consensus is computed.. |
| Sequence Annotation | Per sequence annotation of named attributes and values. Example: Processed sequencing data for submission to dbEST without assembly. Reads have already been submitted to one of the sequence read archives in raw form. The fasta data submitted under this analysis object result from the following treatments, which may serve to filter reads from the raw dataset: - sequencing adapter removal - low quality trimming - poly-A tail removal - strand orientation - contaminant removal. |
| Abundance Measurement | Identify the tools and processing steps used to produce the abundance measurements (coverage tracks). |
Data files for Analysis
Analysis に含めるデータファイルを選択します。
- Run/Analysis
- データファイルが Run もしくは Analysis に属しているのかを指定します。ウェブ画面上では入力できず、属している Run もしくは Analysis の alias が選択されると自動的に入力されます。タブ区切りテキストファイルで入力する場合には、Run もしくは Analysis を入力します。
- File Name*
- 解析データのファイル名。
- Run/Analysis contains files*
- データファイルが属する Analysis を選択します。
- File Type*
- 解析データのファイル形式。
| File Type | Description |
|---|---|
| bam | Binary form of the Sequence alignment/map format for read placements, from the SAM tools project. See http://sourceforge.net/projects/samtools/. |
| tab | A tab delimited text file that can be viewed as a spreadsheet. The first line should contain column headers.. |
| ace | Multiple alignment file output from the phred assembler and similar programs. See http://www.phrap.org/consed/distributions/README.16.0.txt for a description of the ACE file format.. |
| fasta | Sequence data format indicating sequence base calls.The format is simple: a header line initiated with the > character, data lines following with base calls.. |
| wig | The wiggle (WIG) format allows display of continuous-valued data in track format.This display type is useful for GC percent, probability scores, and transcriptome data. See http://genome.ucsc.edu/goldenPath/help/wiggle.html for a description of the Wiggle Track format.. |
| bed | BED format provides a flexible way to define the data lines that are displayed in an annotation track. See http://genome.ucsc.edu/FAQ/FAQformat#format1 for a description of the BED format.. |
| vcf | Variant Call Format. See http://www.1000genomes.org/wiki/analysis/variant%20call%20format/vcf-variant-call-format-version-41 for a description of the VCF format. |
| maf | Mutation Annotation Format |
| gff | General Feature Format |
| csv | |
| tsv |
- MD5 Checksum*
- Analysis データファイルの MD5 チェックサム値。MD5 チェックサム値の取得方法