Pipeline(サービス終了)
Basic Analysis(Preprocessing/Mapping/de novo Assembly)
<注意事項>
- 問題がございましたらpipeline_dev@ddbj.nig.ac.jpまでお問い合わせください。
- Error終了したJOBは、順番にpipelineチームが原因を解析しております。
対処策をメールで連絡させて頂く事がございます。 - JOB投入数の制限は、外しました。
- FAQはこちらです。
- DDBJing 講習会(2013.7.4)資料
基礎解析部では以下の処理を行います。
それぞれ画面遷移が異なりますので表の「順」と「画面」でご確認ください。
Preprocessing
fastq形式ファイルのQSを各種グラフで参照できます。fastq形式ファイルの編集ができます。
<注意事項>
ファイルをアップロードする場合、paired-endでは、ファイル名を拡張子の直前で、_1, _2 として区別して下さい。(
例:test_data_1.fastq , test_data_2.fastq )
| 順 | 画面 | 項目 |
|---|---|---|
| 1 | LOGIN | User ID, Password |
| Registration form | User ID, Email address, First name, Last name, Institution with department, Country, Address, Postal/Zip code, Telephone number, Purpose of utilizaion | |
| 2 | Selecting Query Files | query files(fasta/fastq) |
| *FTP Uploadの場合、Registration of fasta/fastq files | read layout(single-end/paired-end)選択, Instrument model選択, Study title | |
| 3 | Set Perameters for Preprocessing | query用fastqファイル編集(QSによるトリム及び、各種条件でのリード除去) |
| 4 | Run Confirmation | Email address |
| 5 | Status - Preprocessing/Mapping/de novo Assembly | 実行ジョブstatus確認 |
| 6 | Detail view | fastqファイル, QS Average(PDF), QS Count(PDF), QS Error(PDF) |
- + fastqファイル
- read編集(指定QS未満をトリム他、各種条件による選別)、片側のみのpaired-endリード削除
- + QS Average(PDF), QS Count(PDF)
- 編集前のQSの平均と標準偏差を計算、グラフ作成
- + QS Error(PDF)
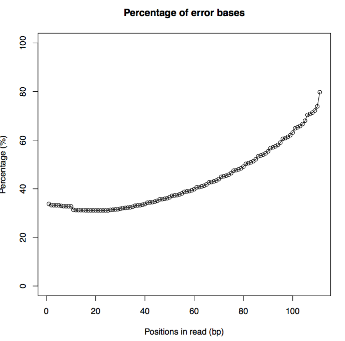
- 編集後のリード位置毎の削除割合を計算、グラフ作成
Preprocessing処理後のファイルは、「Preprocessingタブ(下図黄色ハイライト)」でMapping/de novo Assemblyのクエリとして選択できるようになります。クエリファイルの先頭は、JOB番号です(下では3845がJOB番号)。
Mapping (Reference Alignment)
| 順 | 画面 | 項目 |
|---|---|---|
| 1 | LOGIN | User ID, Password |
| Registration form | User ID, Email address, First name, Last name, Institution with department, Country, Address, Postal/Zip code, Telephone number, Purpose of utilizaion | |
| 2 | Selecting Query Files | query files(fasta/fastq) |
| *FTP Uploadの場合、Registration of fasta/fastq files | read layout(single-end/paired-end)選択, Instrument model選択, Study title | |
| 3 | Selecting Tools for Basic analysis of DDBJ ANNOTAION PIPELINE | tool選択 |
| 4 | Generating Query Sets from Query Read Files | query用fastqファイルのreadファイル選択 |
| 5 | Specifying Database of Reference Genome | reference genomeのセット |
| 6 | Set Options | tool毎のoption, ‘uniq’選択, DNA polymorphism抽出方法選択 |
| 7 | Run Confirmation | Email address |
| 8 | Status - Preprocessing/Mapping/de novo Assembly | 実行ジョブstatus確認 |
| 9 | Detail view | Error Rate, Coverage, Depth, Map ratio, コマンド毎の結果ファイル(samフォーマット) |
- + ErrorRate (mapping, graph)
- Percentage error of mapped sequence to reference sequence is calculated by read position.
- + Coverage (mapping, numeric data)
- Sum of the length of all contigs/G,
where
G = Size (bp) of Reference Genome excluding “N” nucleotides L = Sequence Length (bp),
N = # sequences. - + Depth (mapping, numeric data)
- The average of total sequence length (length of all sequence reads
in a contig including gaps)/contig
Length excluding “N” nucleotides.
Reference: Lander ES, Waterman MS, Genomic mapping by fingerprinting random
clones: a mathematical analysis.
Genomics 1988, 2(3):231-239. - + Map ratio (mapping, numeric data)
- Number of mapped reads* / Number of reads
*: the number of reads, which were mapped in both ends.
de novo Assembly
| 順 | 画面 | 項目 |
|---|---|---|
| 1 | LOGIN | User ID, Password |
| Registration form | User ID, Email address, First name, Last name, Institution with department, Country, Address, Postal/Zip code, Telephone number, Purpose of utilizaion | |
| 2 | Selecting Query Files | query files(fasta/fastq) |
| *FTP Uploadの場合、Registration of fasta/fastq files | read layout(single-end/paired-end)選択, Instrument model選択, Study title | |
| 3 | Selecting Tools for Basic analysis of DDBJ ANNOTAION PIPELINE | tool選択 |
| 4 | Generating Query Sets from Query Read Files | query用fastqファイルのreadファイル選択 |
| 5 | Set Options | tool毎のoption, ‘uniq’選択, DNA polymorphism抽出方法選択 |
| 6 | Run Confirmation | Email address |
| 7 | Status - Preprocessing/Mapping/de novo Assembly | 実行ジョブstatus確認 |
| 8 | Detail view | Contig数, Total contig size, Maximum contig size, Minimum contig size, N50 contig size, コマンド毎の結果ファイル (samフォーマット) |
LOGIN
- システム利用に際してのアナウンスは、”twitter”で行ってます。
- PipelineのIDをお持ちでない場合、「新規アカウント作成」で、登録画面へ遷移します。
- 試験的に使用したい方は「”guest”としてログイン」で、デモ画面を確認できます。
- “動作中JOBの確認”では、”guest”としてStatus画面へ遷移し、JOBの実行状況が確認できます。
- マニュアルおよびチュートリアルが用意されています。
- DRAアカウント登録はこちらのページです。 please see the page.
Registration form
アカウント登録
<注意事項>
- スーパーコンピュータの新システム移行に伴い、既にアカウントをお持ちの方も登録内容に追加項目があります。
- ②に該当する項目をAdditional input(追加項目ページ)から入力します。
- 登録が完了すると、User ID, Initial passwordが、Email address宛に自動配信されます。
- Email addressの記入には十分注意して下さい。
- 新システム移行に伴い、新たに追加した項目です。
- 全ての記入項目を確認後、登録します。
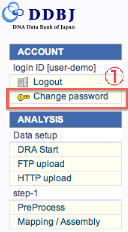
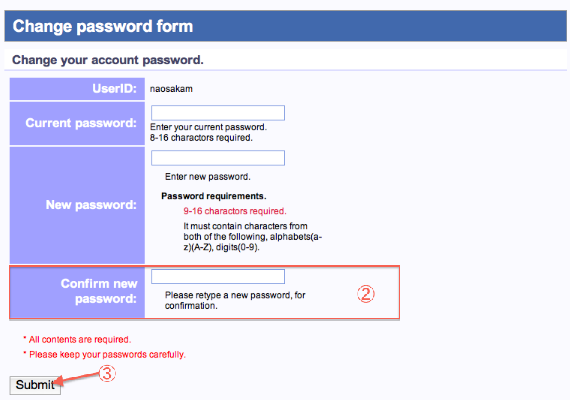
パスワードの変更
- passwordの変更は、各画面の左側メニュー (Change password) からいつでも行えます。
- パスワードは確認の為、再入力します。
- 全ての記入項目を確認後、実行します。
Selecting Query Files
- DRA(DDBJ Sequence Read Archive)に登録したデータ
- HTTPでのアップロード(新規、既存ファイル)
- FTPでのアップロード(新規、既存ファイル)
- DRA databaseからDRA/ERA/SRA のFASTQファイルをインポート
- Preprocessing処理での結果ファイル
- BWA(mapping tool)でのUnmap結果ファイル
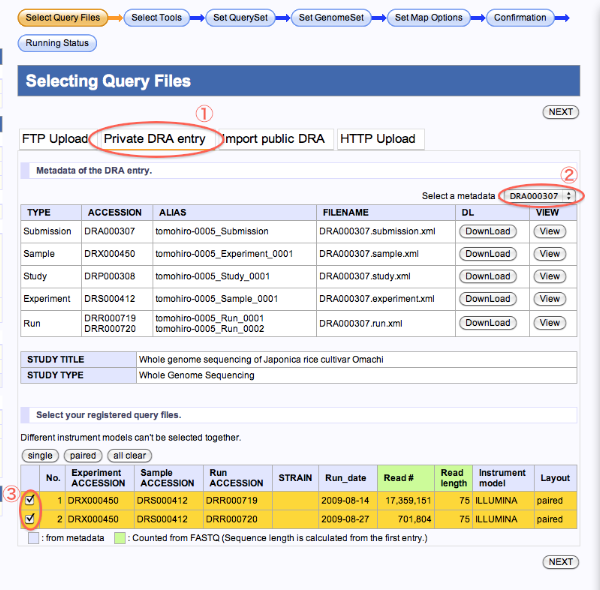
DRA(DDBJ Sequence Read Archive)に登録したデータ
- Private DRA entryを選択します。
- metadataを選択します。
- Queryにするファイルを選択します。
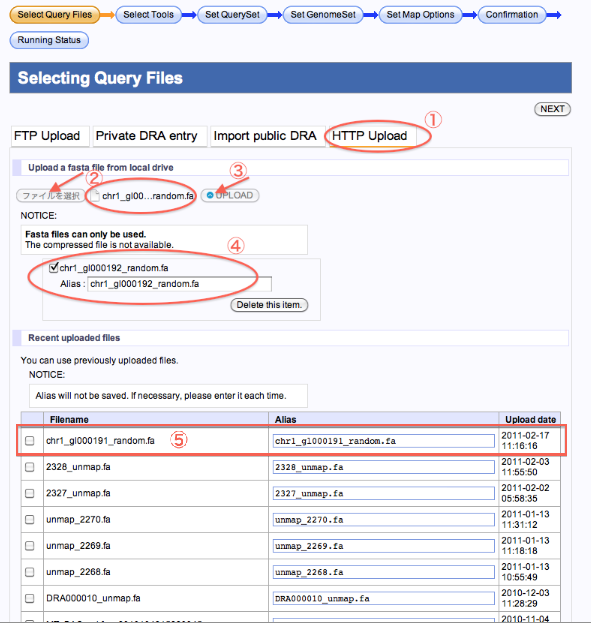
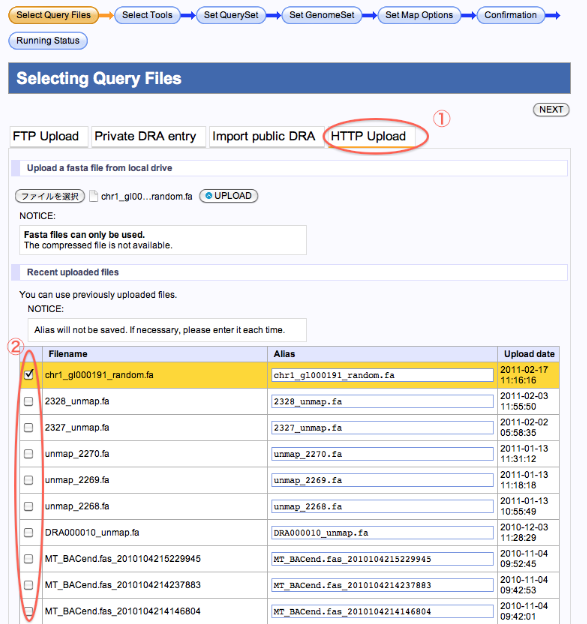
HTTPでのアップロード(新規、既存ファイル)
新規にHTTPアップロードする場合
- HTTP Uploadを選択します。
- ”ファイルを選択”をクリックしローカルからファイルを選択します。
- ”UPLOAD”をクリックします。
- ファイルアップロードが完了するとファイル名が表示されます。Aliasを入力できます。
- リロードすると表の中にファイルが表示されています。
既にHTTPアップロードしたファイルから選択する場合
- HTTP Uploadを選択します。
- 既にアップロード済みのファイルから選択します。
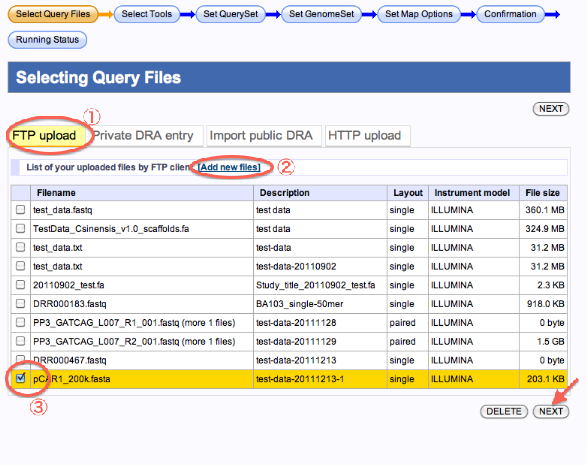
FTPでのアップロード(新規、既存ファイル)
- FTP Uploadを選択します。
- 新規にファイルをアップロードする場合は、[Add new files]をクリックします。
—–>Registration of fastq/fasta files画面へ遷移します。 - 既にFTPアップロードしたファイルを使用する時は、リストから選びます。
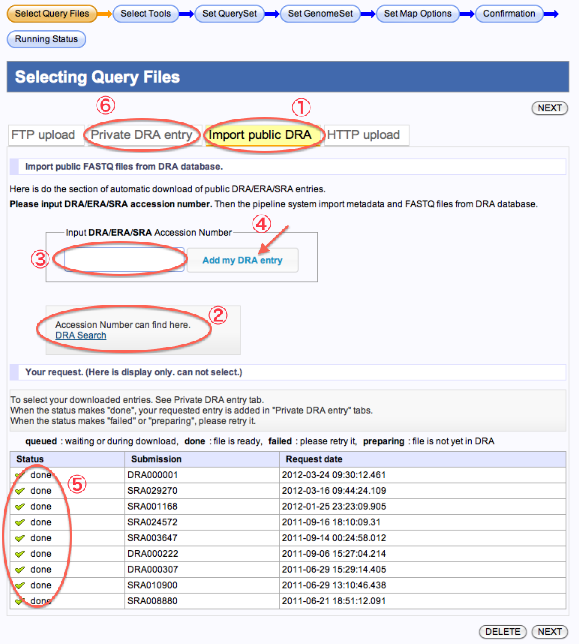
DRA databaseからDRA/ERA/SRA のFASTQファイルをインポート
- Import public DRAを選択します。
- Accession Numberを検索したい場合はこちらからできます。
- Accession Numberを入力します。
- Add my DRA entry をクリックします。
- インポートが終了すると、Statusが”queued”から”done”に変わります。(ページ再読み込み)
- Private DRA entryを選択して下さい。インポートしたデータが使用可能となっております。
- インポートが終了するとメールが届きます。
- Statusが”failed”の時は、再実行してください。
- Statusが”preparing”の時は、まだDRAにファイルが準備されておりません。後日、再実行してください。
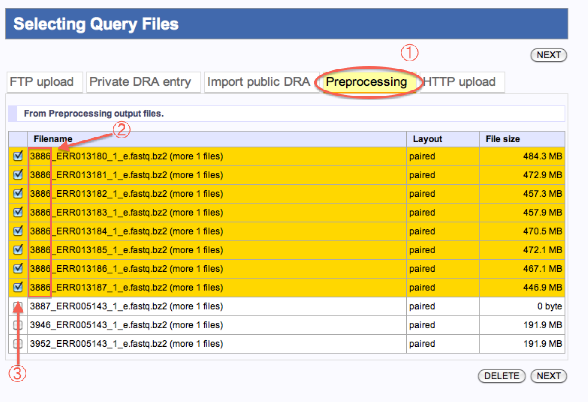
Preprocessing処理での結果ファイル
- Preprocessingを選択します。
- Preprocessing結果ファイルは、「JOB番号_ファイル名_e」で表示されています。
(BWA Unmap結果ファイルは、「JOB番号_ファイル名.unmapped」で表示されています。 - 使用するファイルをチェックします。
Registration of fastq/fasta files
新規にFTPでFASTA/FASTQファイルをアップロードする方法
By FTP(Recommended)
- 1. Upload FASTA/FASTQ files
- FTPクライアントによる転送方法については、こちらのページをご参照ください。
- FTP clientをローカルPCにインストールし、DDBJのサーバーへFTP転送します。
- FTP setting内容です。(loginできない場合、パスワード変更を行って下さい。)
- (FTPでの転送ができない場合、時間がかかりますがHTTPでの転送も可能です。)
- アップロードが終了したら、画面をリロードしてください。下のリストにファイルが追加されます。
- アップロードしたファイルをチェックし、次へ進みます。
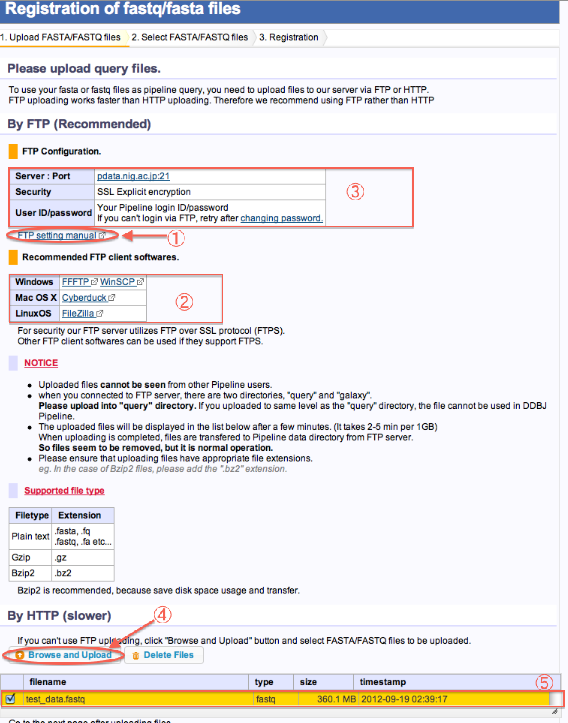
- 2. Select a FASTA/FASTQ file(Uploadしたファイルの注釈付け1)
- Read layoutでSingle-end又は、Paired-endを選択します。
- read fileを選択します。(paired_endの場合はread1と対になるread2も選択)
- 次へ進みます。
<Single_endの場合>
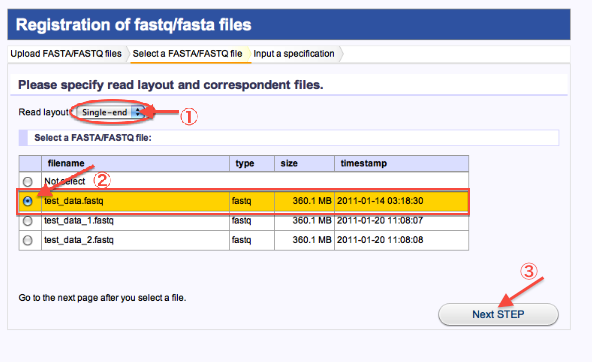
<Paired_endの場合>
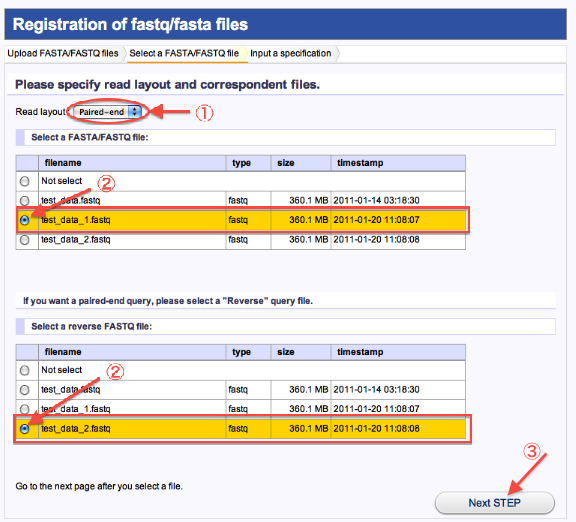
- 3. Input a specification(Uploadしたファイルの注釈付け2)
- シークエンサの機種を選択します。
- Study titleを入力します。
- 登録(SUBMITをクリック)します。
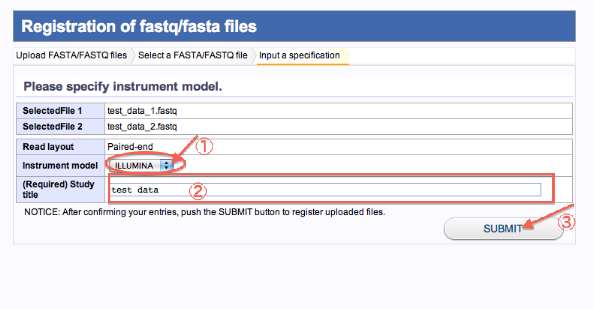
- 処理終了、Assembly/Mapplingをクリックすると、Selecting Query Files画面に遷移します。
*Uploadしたファイルを使用して解析が可能になっています。
By HTTP(slower)
- Browse and Uploadをクリックします。
- ローカルPCからファイルを選択します。開始するとUpload経過が表示されます。
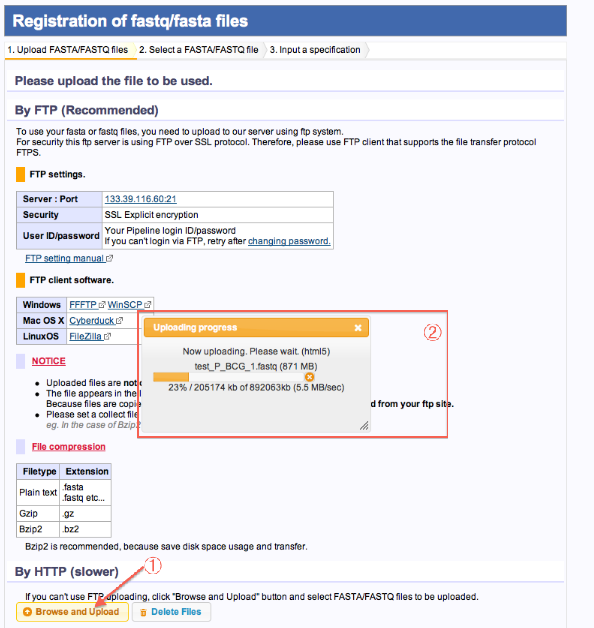
- Uploadが完了したらページ再読み込みします。
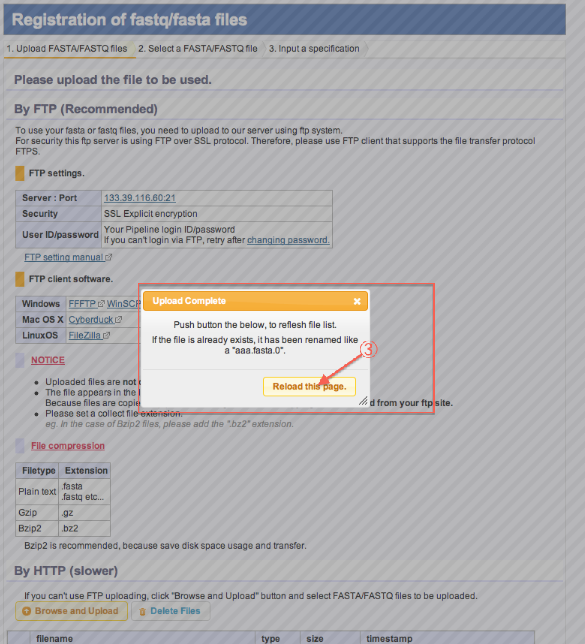
- ファイルがリストに追加されています。
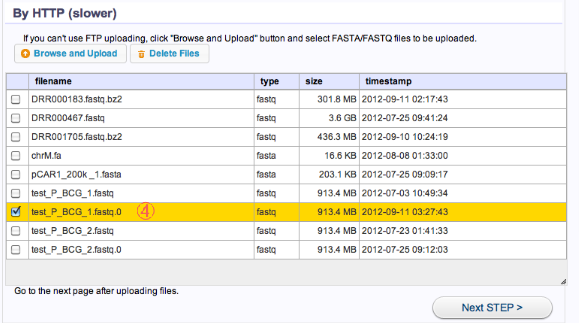
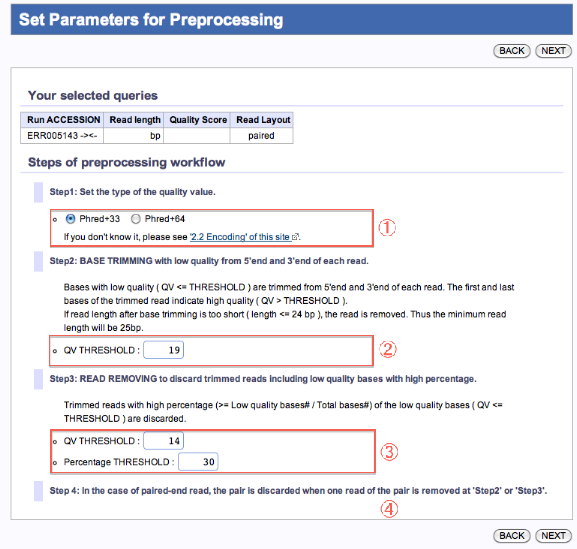
Set Parameters for Preprocessing
Preprocessing処理によるFastq形式ファイルの編集
- QVタイプを選択します。(参照:2.2 Encoding)
- 5’, 3’両端から、「指定QV」より大きい値の塩基が出現するまでトリムします。
(トリム後のリード長が24bp以下の場合、そのリードを取り除きます。) - 「指定QV」未満の塩基が、トリム後のリード長の「指定%」より多い場合は、そのリードを取り除きます。
- ペアードエンドリードの場合、片方が条件 ② 、③ により取り除かれた場合、もう一方も取り除かれます。
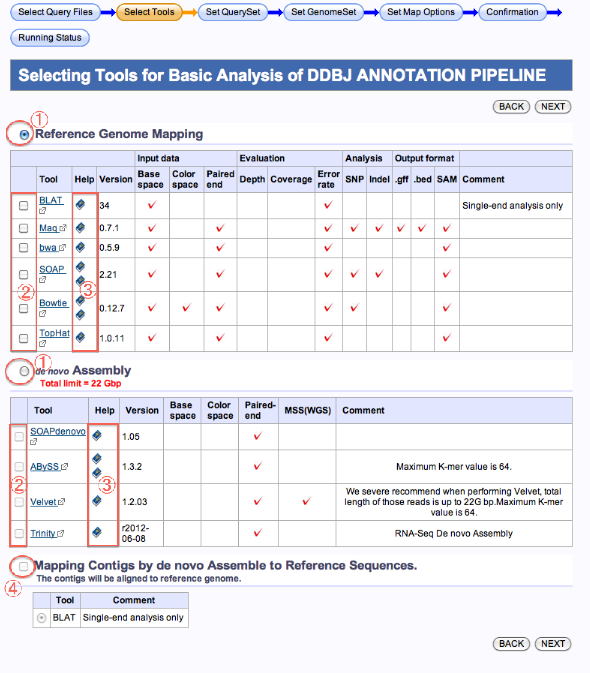
Selecting Tools for Basic analysis of DDBJ ANNOTATION PIPELINE
解析ツールの選択
- まず最初に処理 ( Reference Genome Mapping または、de novo Assembly ) を選択します。
- ツールを選択します。
- この後でオプションの指定等あります。
ツールのマニュアル(Help列:本マーク)をよくお読み下さい。 - de novo Assembly の場合で、結果contigをqueryとして、続けて Mapping (BLAT使用)する場は、下の、Mapping Contigs by de novo Assemble to Reference Sequences.を選択します。
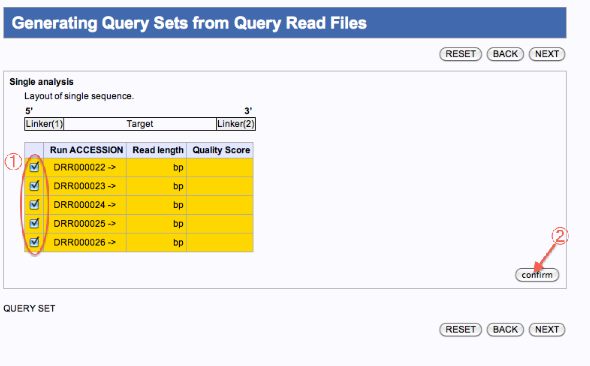
Generating Query Sets from Query Read Files
クエリーファイルを一つのジョブで実行
- 編集したいファイルにチェックを入れます。
- confirmをクリックします。
- 確認
- 次へ
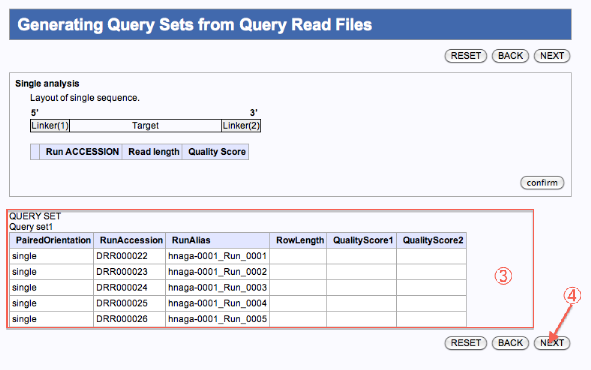
クエリーファイルを複数のジョブで実行
- 一つのジョブとして編集したいファイルにチェックを入れます。
- confirmをクリックします。
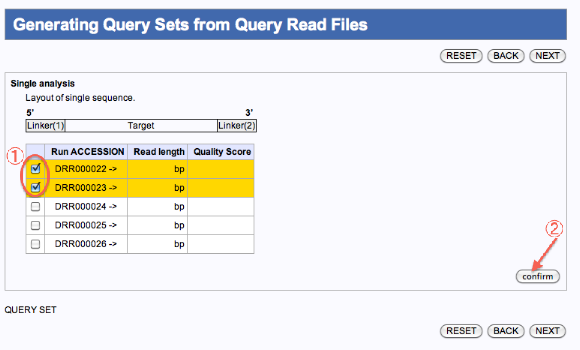
- 確認
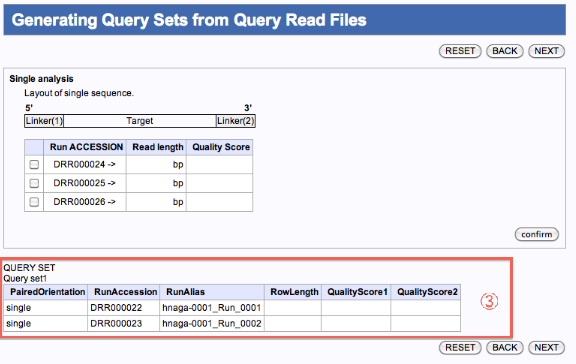
- 残りのファイルの内、別のジョブとして編集したいファイルにチェックを入れます。
- confirmをクリックします。
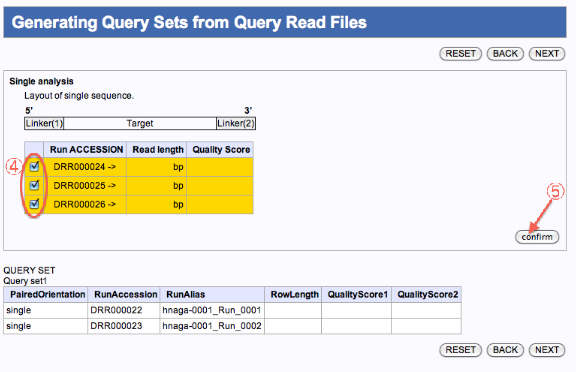
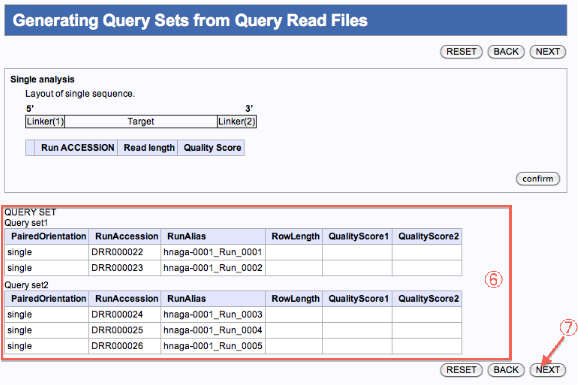
- クエリセット1、2ができます。(JOBが2つ投入)
- 次へ
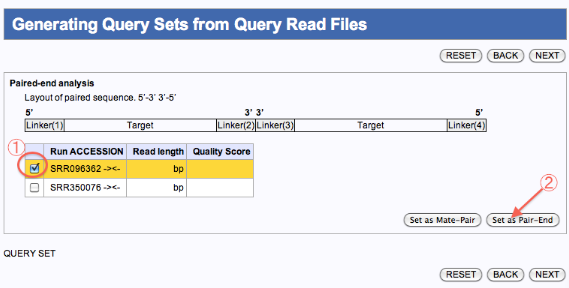
Mate-pairedを使用してdenovoAssembly(SOAPdenovo,Velvetのみ)
- 編集したいファイルにチェックを入れます。
- 先にPair-Endをセットします。(その後Mate-Pairをセットします)
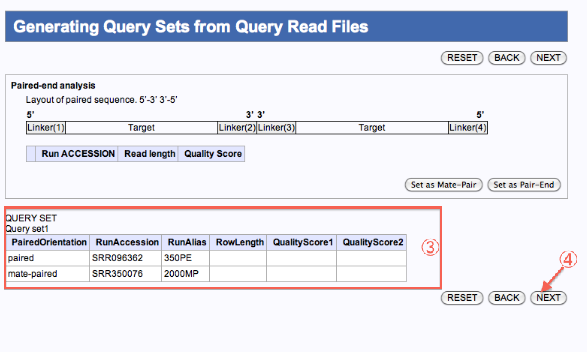
- 確認
- 次へ
Specifying Database of Reference Genome
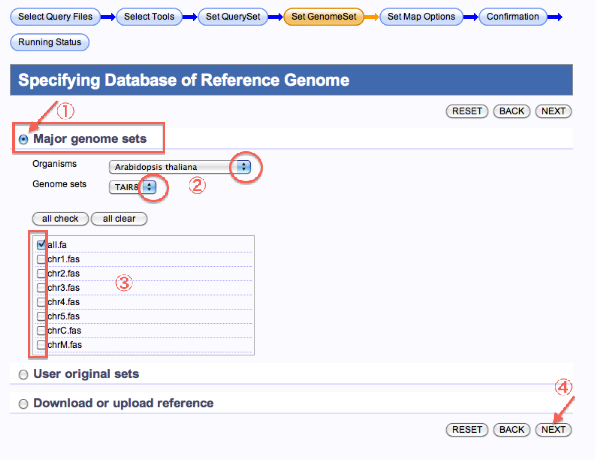
Major genomeとして登録されているreferenceを使用する場合
- Major genome setsを選択します。
- Organisms,Genome setsを選択します。
- 染色体を選択します。
- 次へ
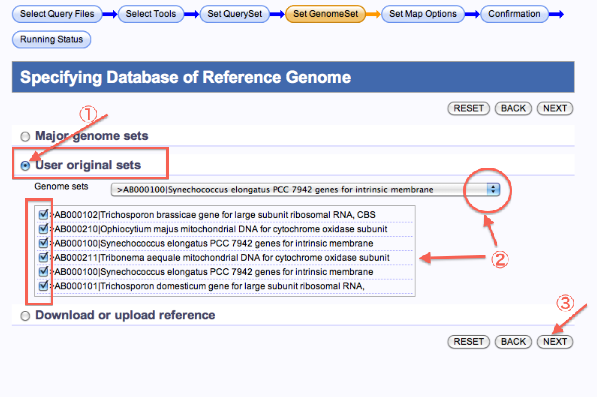
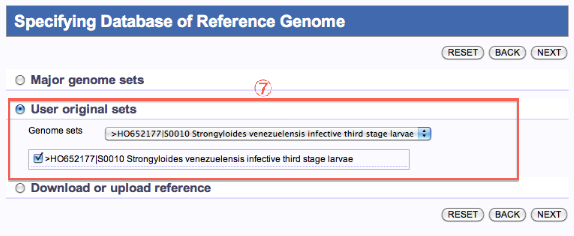
自分で登録したreference (User original sets) を使用する場合
- User original setsを選択します。
- Genome setsを選択します。
- 次へ
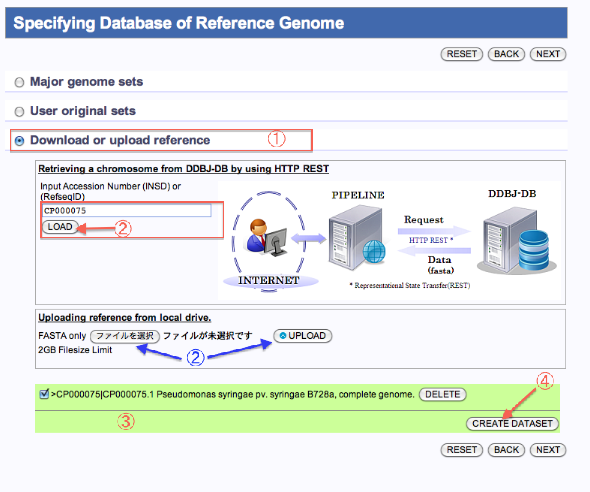
自分でreferenceを(User original setsへ)登録する場合
- Download or upload referenceを選択します。 [アクセッション番号(INSD)からのダウンロードの場合]
- アクセッション番号(INSD)を入れ"LOAD"をクリックします。 [ローカルPCからのアップロードの場合]
- "ファイルを選択"をクリックし、ローカルPCからファイルを選択したら"UPLOAD"をクリックします。
- ファイルが表示されます。
- "CREATE DATASET"をクリックするとCreate Genome Dataset画面へ遷移します。
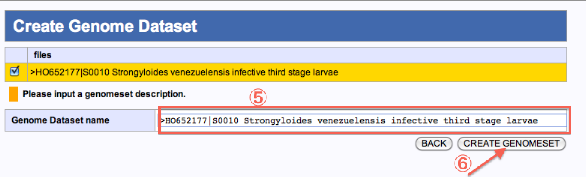
- genomeset の記述を変更できます。
- "CREATE GENOMESET"をクリックするとSpecifying Database of Reference Genome画面に戻ります。
- ダウンロードしたファイルが"User original sets"に追加され、選択した状態となっています。
Set Options
Setting for De Novo Assembly
- オプションを指定します。
- WGS配列データとしてDDBJに登録する場合は該当にチェックします。
- 次へ
*ツールにより、画面は若干異なります。
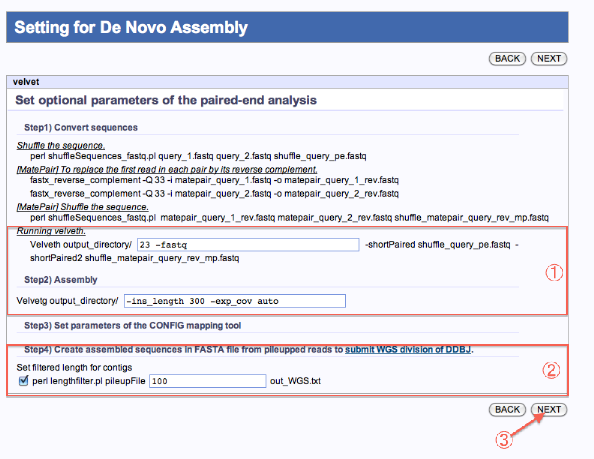
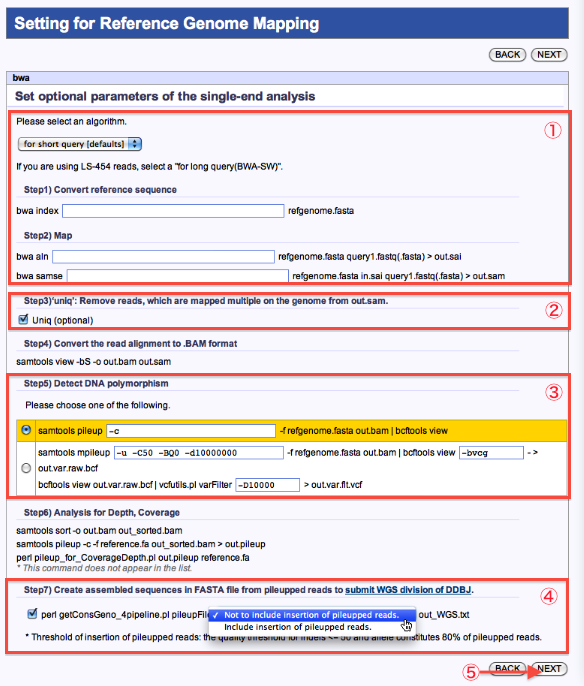
Setting for Reference Genome Mapping
- オプションを指定します。
- ‘Uniq’指定ができます。
- DNA polymorphism抽出方法を選択します。
- WGS配列データとしてDDBJに登録する場合は該当にチェックします。
- 次へ
*ツールにより、画面は若干異なります。
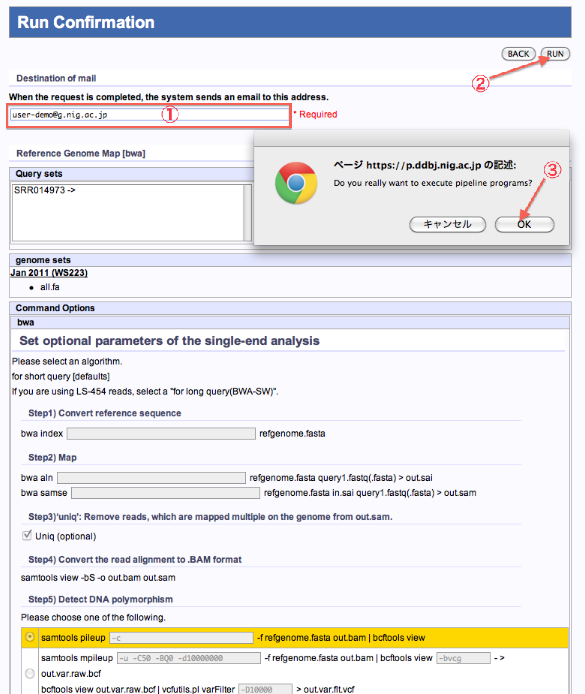
Run Confirmation
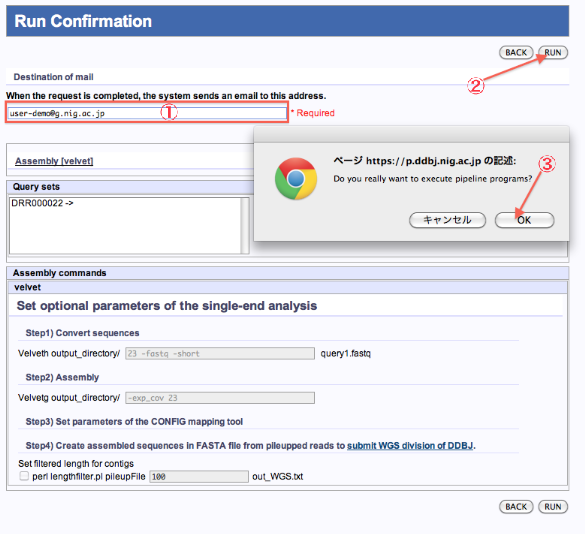
Mapping/de novo Assembly
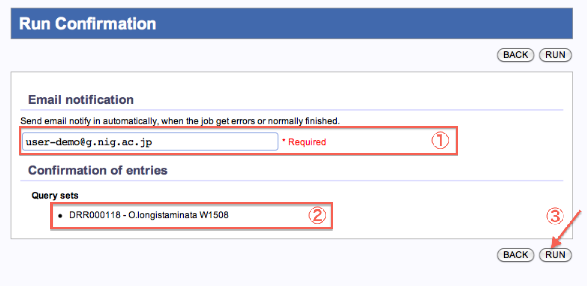
- ジョブが終了した際の連絡メールアドレスを記入します。
- 内容を確認したら、”RUN”をクリックします。
- PopUp表示で再確認します。”OK”で実行。The reservation was completed.画面へ遷移します。
- “STATUS”をクリックすると、Mapping, de novo Assembly 各Status ページへ遷移します。
*guestユーザーでは、RUNボタンが表示されません
<Mapping (tool:bwa)の例>
<de novo Assembly (tool:velvet)の例>
Preprocessing
- ジョブが終了した際の連絡メールアドレスを記入します。
- 内容を確認します。
- “RUN”をクリックすると、The reservation was completed.画面へ遷移します。
- “STATUS”をクリックすると、Status-PreProcess画面へ遷移します。
Status-Mapping/de novo Assembly/PreProcess
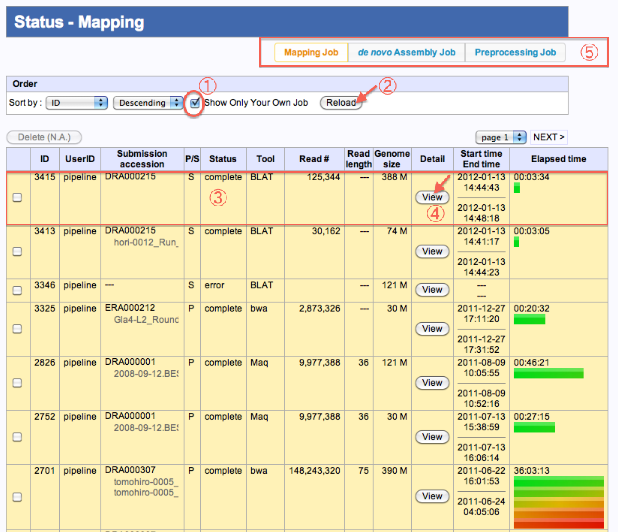
ジョブ実行状況の確認
- ”Show Only Your Own Job”をチェックします。
- Reloadをクリックすると、ログインユーザーの結果のみ抽出されます。
- 実行したジョブのStatusが確認できます。( generating/running/complete/error )
- “View”クリックで、実行状況の詳細画面へ遷移します。
- 他のStatus画面へも遷移できます。
Detail view
Mapping
- 統計結果が表示されます。
- 実行ログの確認ができます。
- 各種コマンド結果ファイルがダウンロードできます。
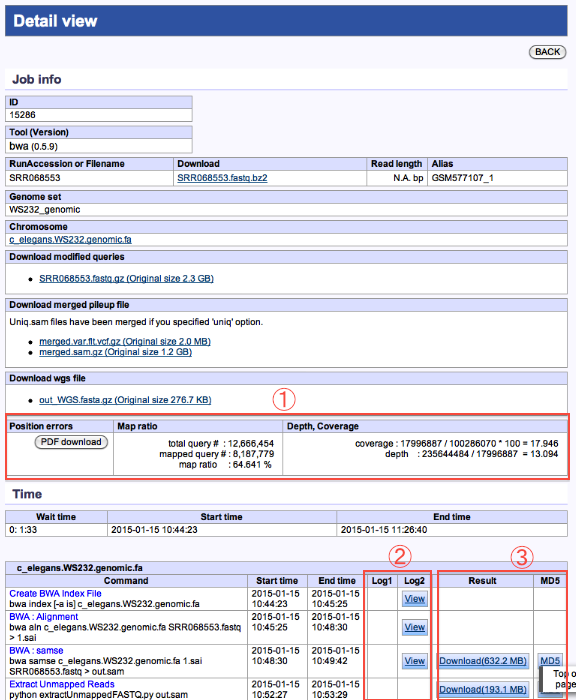
統計結果のダウンロード
- <Position errors>
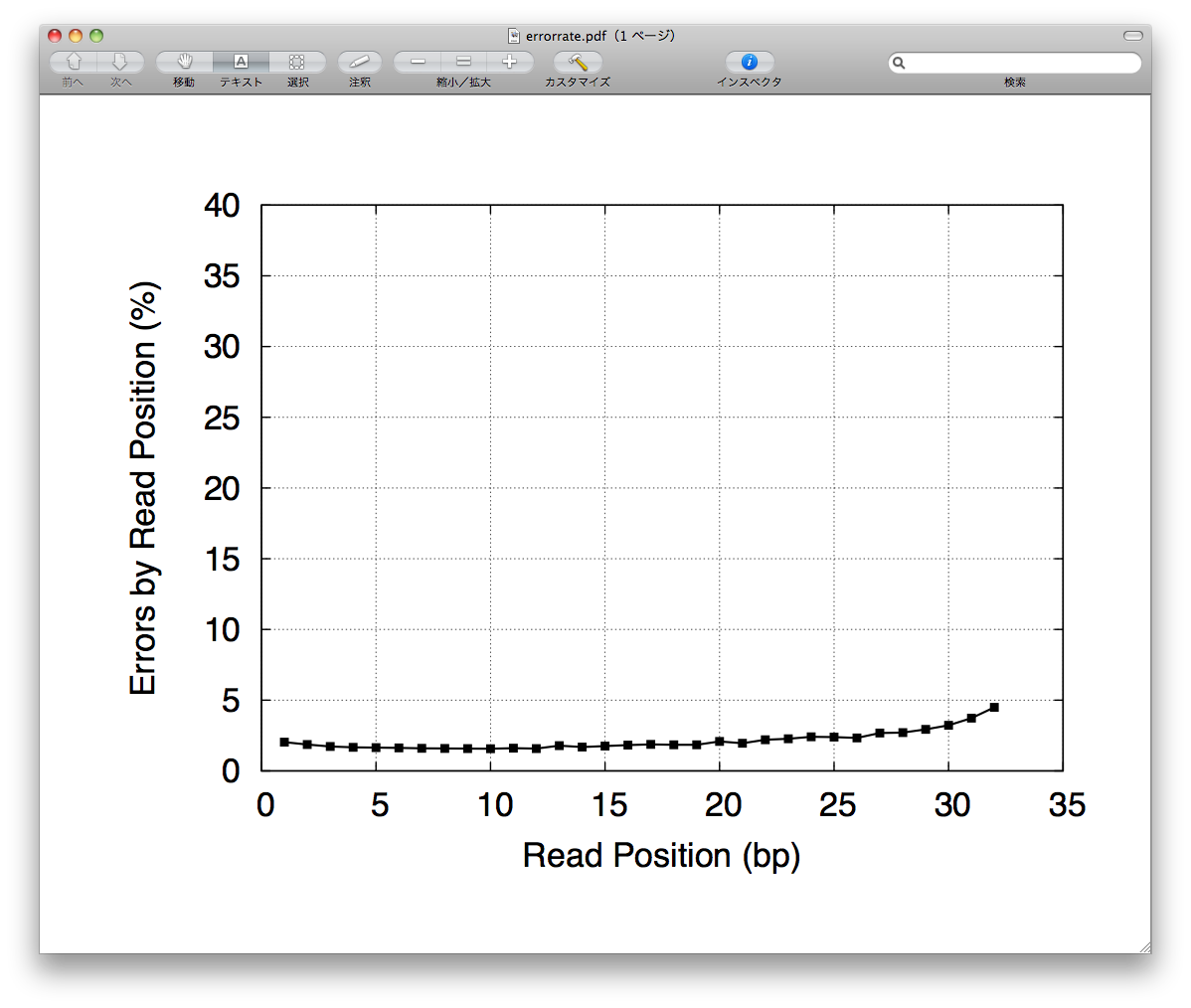
-
- + ErrorRate (mapping, graph).
- Percentage error of mapped sequence to reference sequence is calculated by read position.
- <Map ratio>
-
- + Map ratio (mapping, numeric data)
- Number of mapped reads* / Number of reads
*: the number of reads, which were mapped in both ends.
- <Depth, Coverage>
-
- + Depth (mapping, numeric data)
- he average of total sequence length (length of all sequence reads in a contig including gaps)/contig Length excluding “N” nucleotides.
Reference: Lander ES, Waterman MS, Genomic mapping by fingerprinting random clones: a mathematical analysis.
Genomics 1988, 2(3):231-239.
-
- + Coverage (mapping, numeric data)
- Sum of the length of all contigs/G,
where
G = Size (bp) of Reference Genome excluding “N” nucleotides.
L = Sequence Length (bp),
N = # sequences.
de novo Assembly
- 統計結果が表示されます。
- 実行ログの確認ができます。
- 各種コマンド結果ファイルがダウンロードできます。
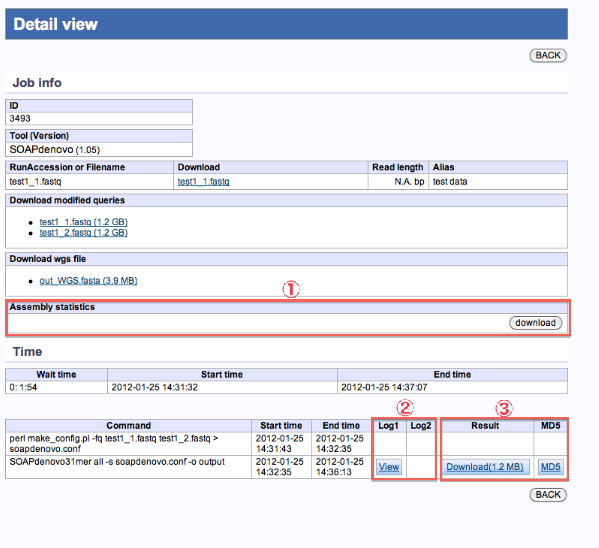
統計量のダウンロード
<de novo Assembly statistics>

Preprocessing
- 編集後Fastqファイル及び、各種グラフのダウンロードが行えます。
- 実行ログの確認ができます。
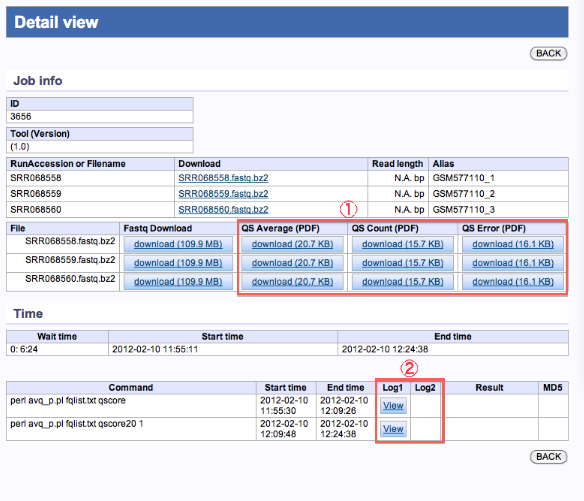
編集済みファイル、各種グラフのダウンロード
- <Fastq Download>
- 編集後のFastqファイルがダウンロードできます。
- <QS Average(PDF)>
- 編集前のQS標準偏差
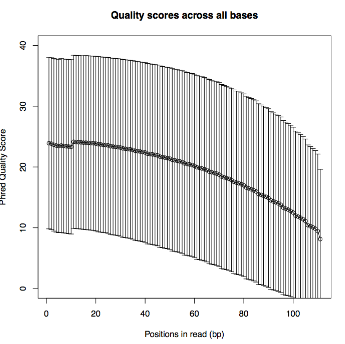
- <QS Count(PDF)>
- 編集前のQS平均
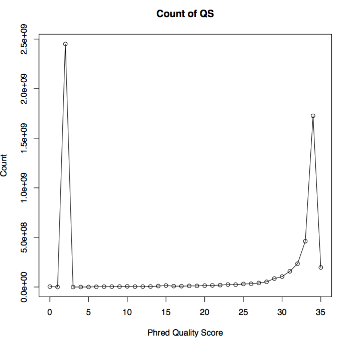
- <QS Error(PDF)>
- 編集後のリード位置毎の削除割合