Sequence Read Archive
Metadata examples
Metadata organization
Submitters can organize metadata objects flexibly.
- Most simple case
- Comparative genome sequencing of three strains (paired-end)
- Biological replicates (paired-end)
- Group submissions by a BioProject
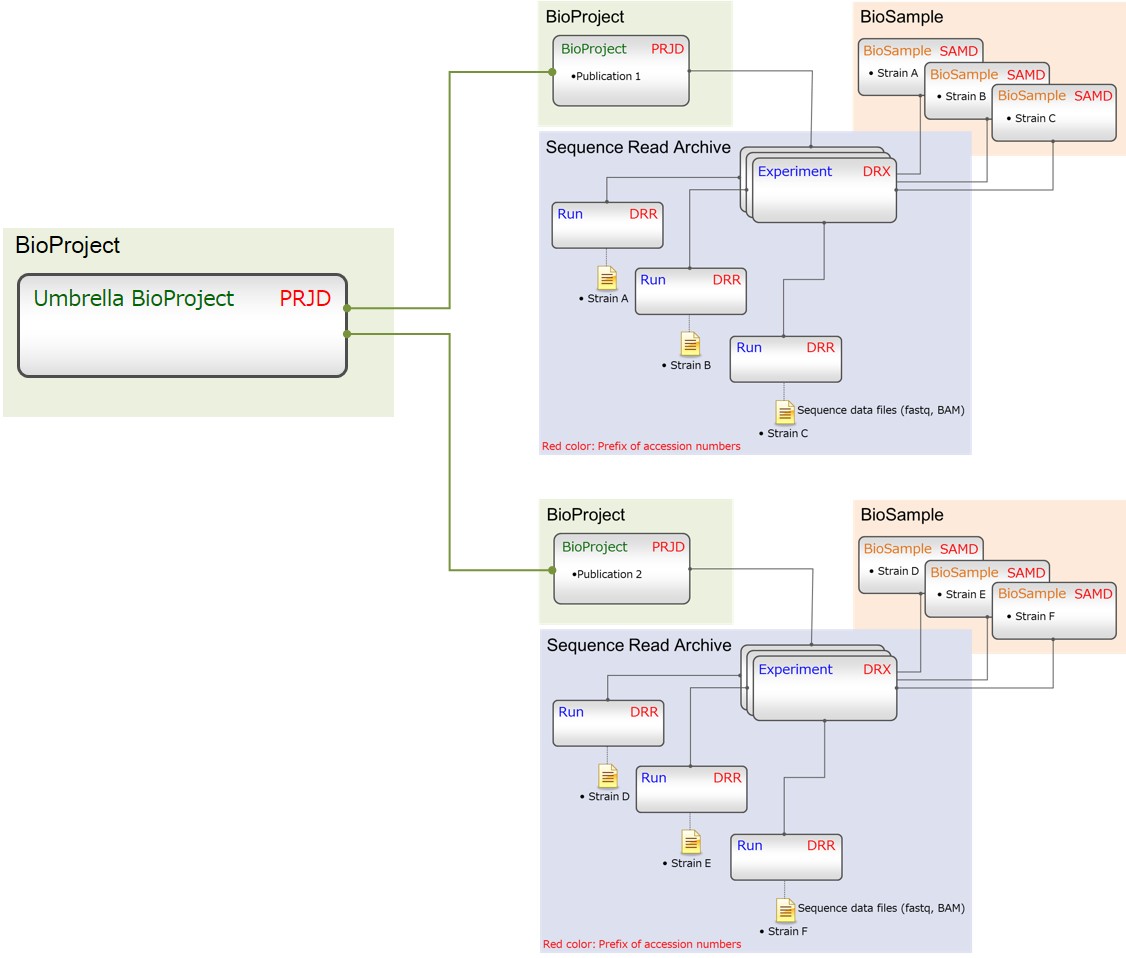
- Group related projects by an umbrella project
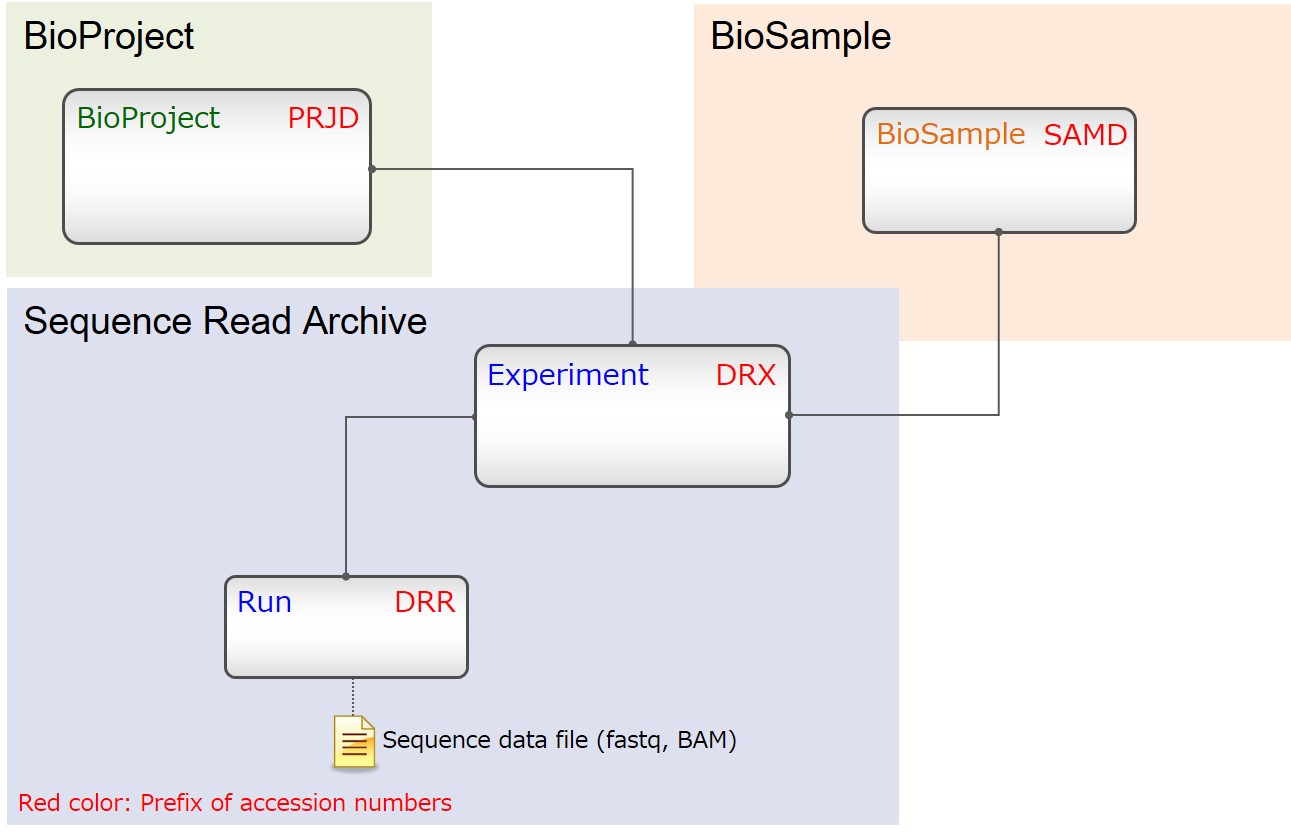
Most simple case
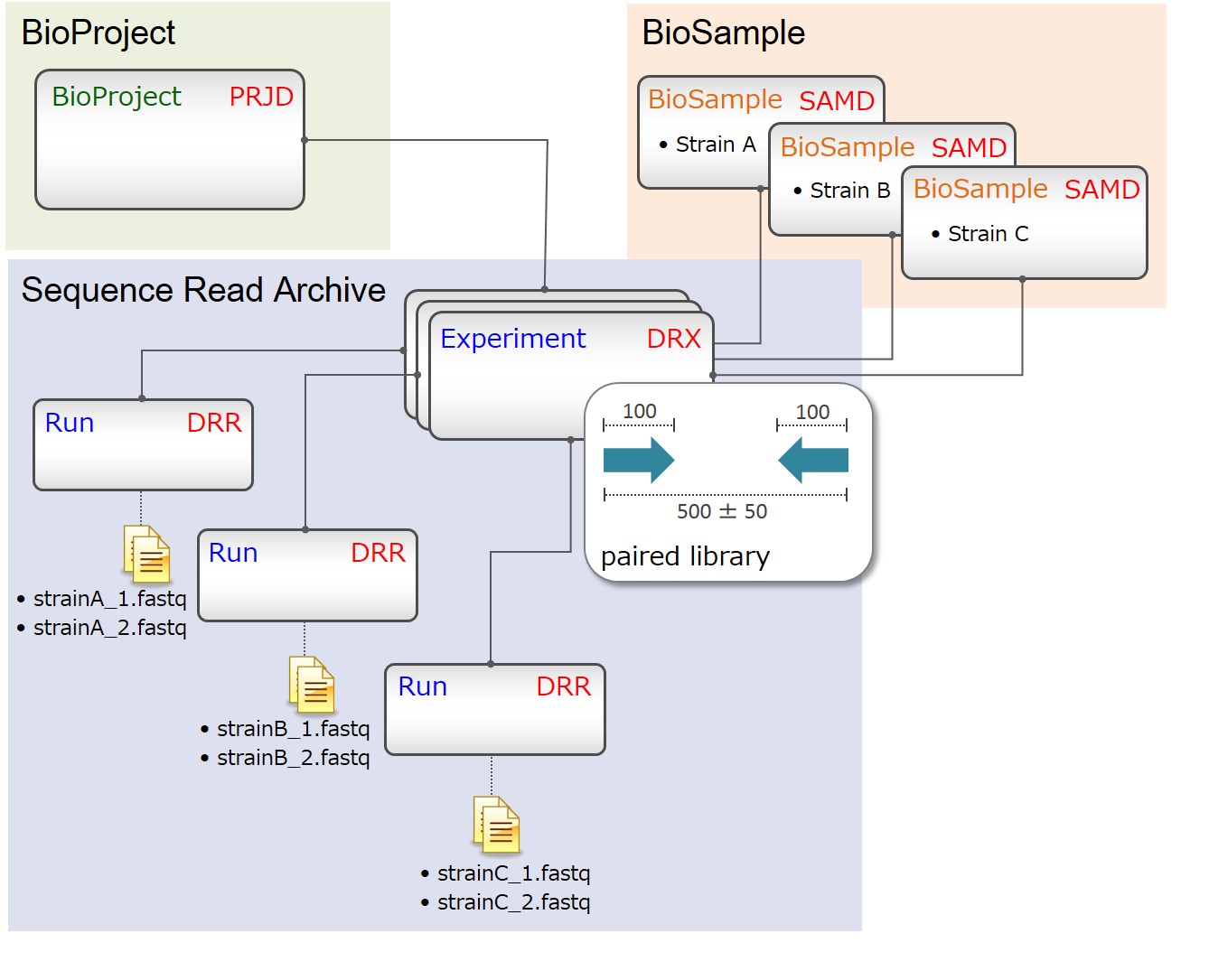
Comparative genome sequencing of three strains (paired-end)
Include paired-end read files in a Run.
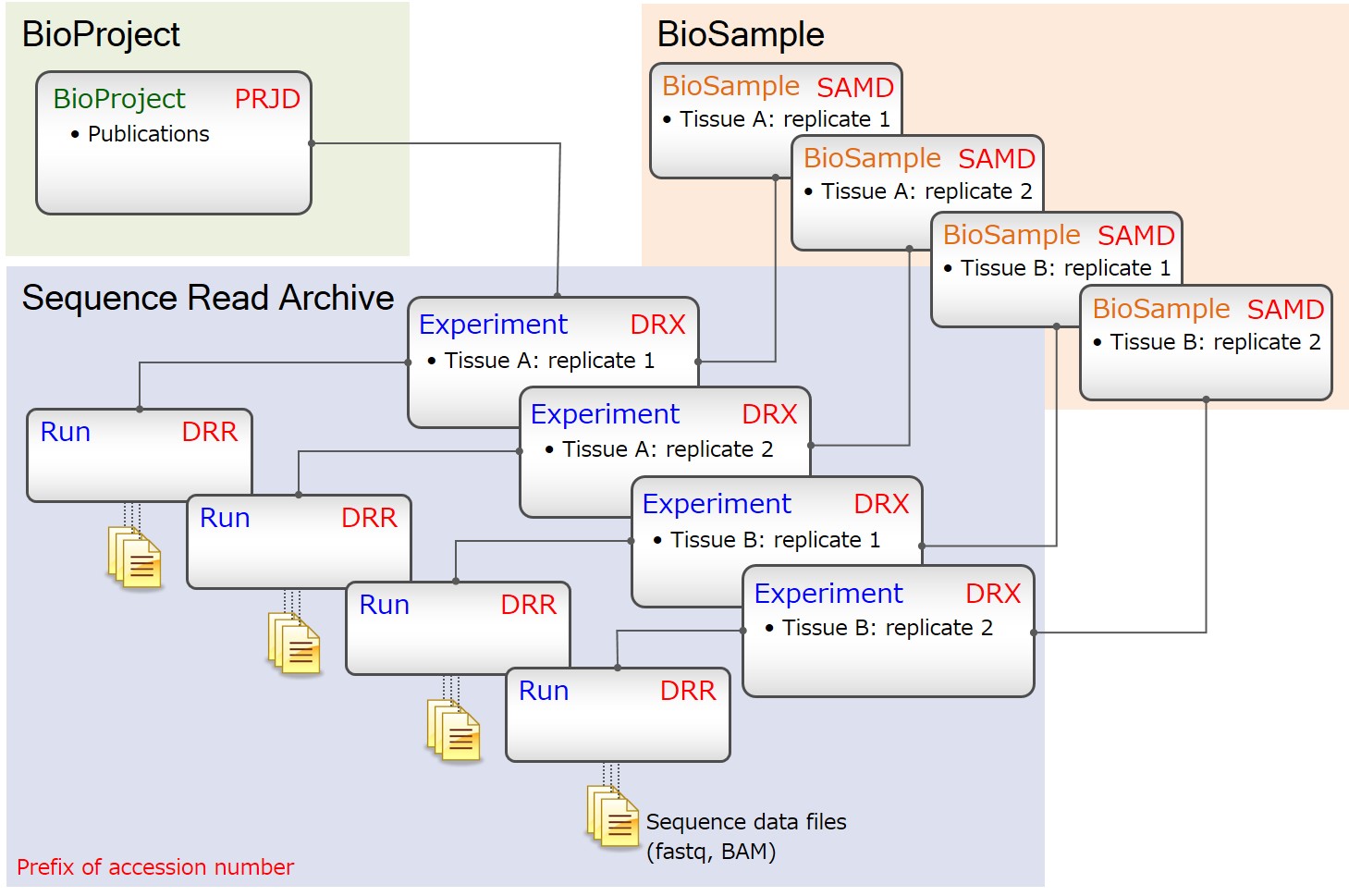
Biological replicates (paired-end)
Group submissions by a BioProject
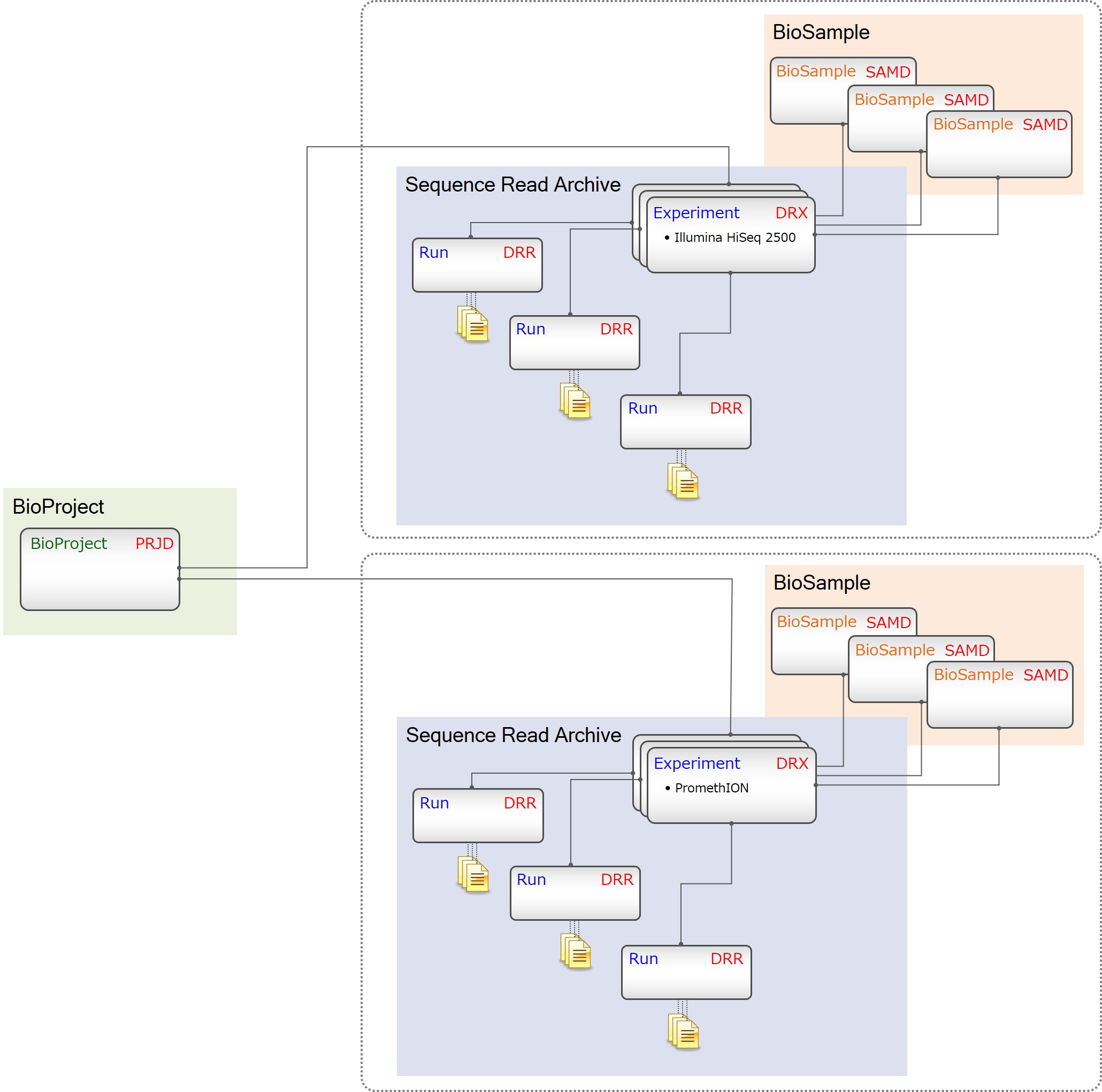
Group related projects by an umbrella project
Metadata tsv example
Example files for submitting Experiment/Run metadata in tab-delimited text (tsv) files.
Example metadata tsv
Metadata excel example
An example metadata excel for submitting Experiment/Run metadata in XMLs generated from the excel.
example-0001_dra_metadata.xlsx