Sequence Read Archive
Metadata
Objects
The metadata describes how the associated data have been obtained. The metadata are composed of 6 objects and each of these objects is defined by its XML schema and is related each other. Metadata examples
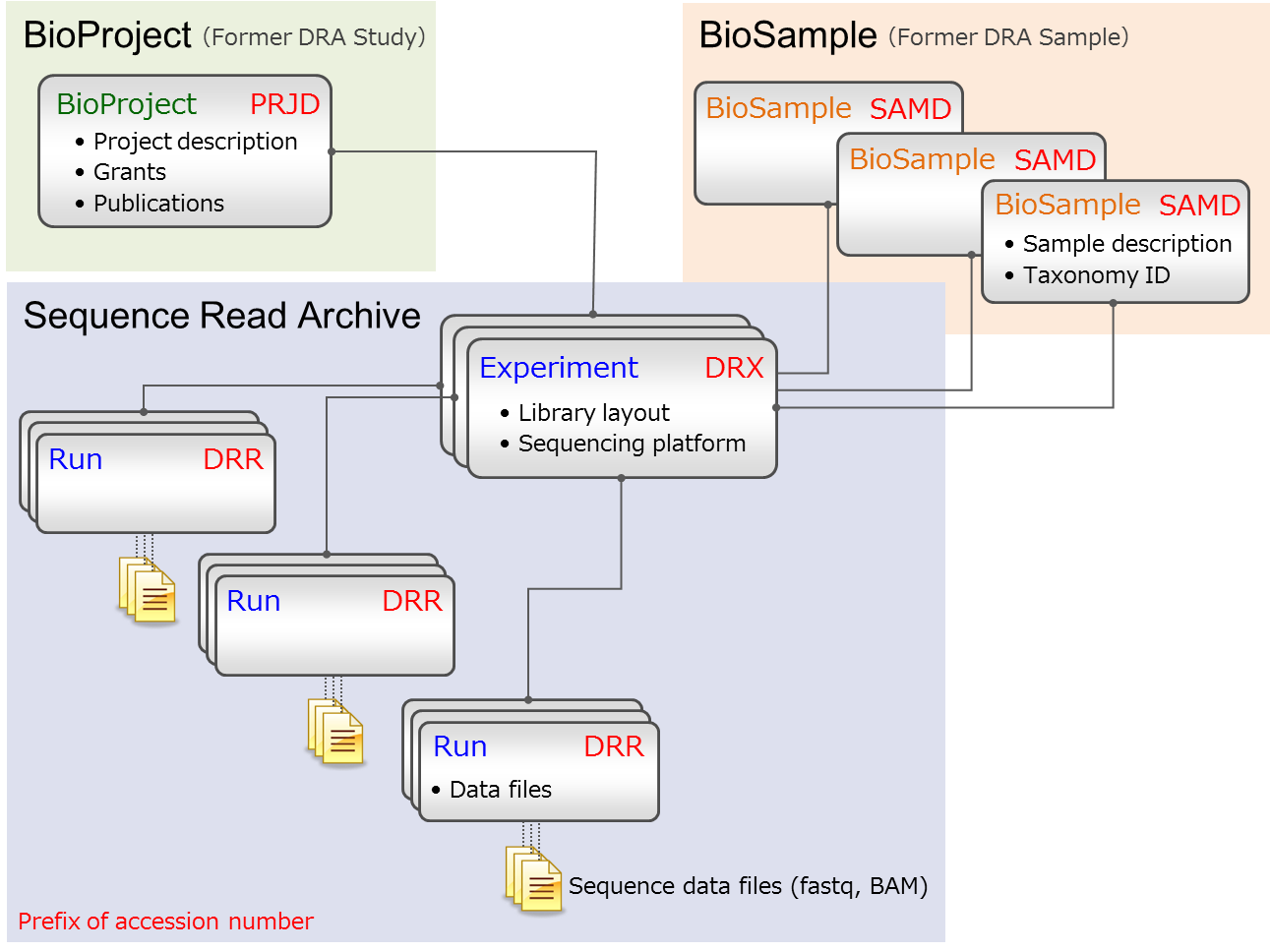
- Submission
- Submission information such as data release and submitters. A container object for grouping objects to be submitted.
- BioProject
- Research project information. External database.
- BioSample
- Biological sample from which sequencing data were obtained. External database.
- Experiment
- A description of sequencing library and instrument. An Experiment references 1 BioProject and 1 BioSample. Multiple Experiments can “point” to a single Sample, but not vice-versa.
- Run
- A Run archives data files which belong to the previously created Experiment (library and instrument). Note that all data files listed in a Run will be merged into a single SRA archive file. Paired-end data files must be listed in a single run in order for the two files to be correctly processed as paired-end.
- Analysis
- Packages data associated with sequence read objects which do not have dedicated databases. Analysis date are not shared among NCBI and EBI.
Metadata fields
Required* Conditionally required*
Submission
Submitting organization
The organization of the account is copied as the Submitting organization. At 20th December 2023, the center name and organization abbreviations were discontinued.
Hold Until
Specify how to release the data.
- Hold Until*
- Direct the DRA to release the record on or after the specified date.Submitter can set the hold date for a maximum of 2 years and can change the date before the record is released.
- Immediate Release*
- Direct the DRA to release the record immediately after submission is processed.
Submitter
The DRA contacts the listed address(es) regarding the submission by e-mail. The contact information is not made public.
BioProject
- BioProject ID*
- Select a project registered to BioProject or submit a new project. For submission to BioProject, please refer to the BioProject Submission.
BioSample
- BioSample ID*
- Select samples registered to BioSample or create and submit new samples. For submission to BioSample, please refer to the BioSample Submission.
Experiment
- Alias
- Name of the experiment designated by the archive. This alias is used to reference metadata objects without accession numbers.
- BioSample Used*
- Select the BioSample this experiment uses.
- Title*
- Short text that can be used to call out experiment records in searches or in displays. A title like “[Sequencing Instrument Model] [paired end] sequencing of [BioSample ID]” (for example, “Illumina HiSeq 2000 paired end sequencing of SAMD00025741”) is automatically constructed. To enter user-defined titles, download Experiment metadata into a tab-delimited text file, edit title values and upload it.
- Library Name
- The submitter’s name for this library.
- Library Source*
- The Library Source specifies the type of source material that is being sequenced.
| Library Source | Description |
|---|---|
| GENOMIC | Genomic DNA (includes PCR products from genomic DNA). |
| GENOMIC SINGLE CELL | Genomic DNA from single cell. |
| TRANSCRIPTOMIC | Transcription products or non genomic DNA (EST, cDNA, RT-PCR, screened libraries). |
| TRANSCRIPTOMIC SINGLE CELL | Transcription products or non genomic DNA from single cell. |
| METAGENOMIC | Mixed material from metagenome. |
| METATRANSCRIPTOMIC | Transcription products from community targets. |
| SYNTHETIC | Synthetic DNA. |
| VIRAL RNA | Viral RNA. |
| OTHER | Other, unspecified, or unknown library source material. |
- Library Selection*
- Whether any method was used to select and/or enrich the material being sequenced.
| Library Selection | Description |
|---|---|
| RANDOM | Random shearing only. |
| PCR | Source material was selected by designed primers. |
| RANDOM PCR | Source material was selected by randomly generated primers. |
| RT-PCR | Source material was selected by reverse transcription PCR. |
| HMPR | Hypo-methylated partial restriction digest. |
| MF | Methyl Filtrated. |
| repeat fractionation | Selection for less repetitive (and more gene rich) sequence through Cot filtration (CF) or other fractionation techniques based on DNA kinetics. |
| size fractionation | Physical selection of size appropriate targets. |
| MSLL | Methylation Spanning Linking Library. |
| cDNA | complementary DNA. |
| cDNA_randomPriming | |
| cDNA_oligo_dT | |
| PolyA | PolyA selection or enrichment for messenger RNA (mRNA); should replace cDNA enumeration. |
| Oligo-dT | enrichment of messenger RNA (mRNA) by hybridization to Oligo-dT. |
| Inverse rRNA | depletion of ribosomal RNA by oligo hybridization. |
| ChIP | Chromatin immunoprecipitation. |
| MNase | Micrococcal Nuclease (MNase) digestion. |
| DNAse | Deoxyribonuclease (DNase) digestion. |
| Hybrid Selection | Selection by hybridization in array or solution. |
| Reduced Representation | Reproducible genomic subsets, often generated by restriction fragment size selection, containing a manageable number of loci to facilitate re-sampling. |
| Restriction Digest | DNA fractionation using restriction enzymes. |
| 5-methylcytidine antibody | Selection of methylated DNA fragments using an antibody raised against 5-methylcytosine or 5-methylcytidine (m5C)MBD2 protein methyl-CpG binding domain : Enrichment by methyl-CpG binding domain. |
| MBD2 protein methyl-CpG binding domain | MBD2 protein methyl-CpG binding domain. |
| CAGE | Cap-analysis gene expression. |
| RACE | Rapid Amplification of cDNA Ends. |
| MDA | multiple displacement amplification. |
| padlock probes capture method | Padlock Probes capture strategy to be used in conjuction with Bisulfite-Seq. |
| other | Other library enrichment, screening, or selection process. |
| unspecified | Library enrichment, screening, or selection is not specified. |
- Library Strategy*
- Sequencing technique intended for this library.
| Library Strategy | Description |
|---|---|
| WGS | Whole genome shotgun. |
| WGA | Whole genome amplification. |
| WXS | Random sequencing of exonic regions selected from the genome. |
| RNA-Seq | Random sequencing of whole transcriptome. |
| miRNA-Seq | Micro RNA and other small non-coding RNA sequencing. |
| ncRNA-Seq | Capture of other non-coding RNA types, including post-translation modification types such as snRNA (small nuclear RNA) or snoRNA (small nucleolar RNA), or expression regulation types such as siRNA (small interfering RNA) or piRNA/piwi/RNA (piwi-interacting RNA). |
| ssRNA-seq | strand-specific RNA sequencing |
| WCS | Whole chromosome (or other replicon) shotgun. |
| CLONE | Genomic clone based (hierarchical) sequencing. |
| POOLCLONE | Shotgun of pooled clones (usually BACs and Fosmids). |
| AMPLICON | Sequencing of overlapping or distinct PCR or RT-PCR products. |
| CLONEEND | Clone end (5’, 3’, or both) sequencing. |
| FINISHING | Sequencing intended to finish (close) gaps in existing coverage. |
| RAD-Seq | Restriction Site Associated DNA Sequence |
| ChIP-Seq | Direct sequencing of chromatin immunoprecipitates. |
| MNase-Seq | Direct sequencing following MNase digestion. |
| DNase-Hypersensitivity | Sequencing of hypersensitive sites, or segments of open chromatin that are more readily cleaved by DNaseI. |
| Bisulfite-Seq | Sequencing following treatment of DNA with bisulfite to convert cytosine residues to uracil depending on methylation status. |
| EST | Single pass sequencing of cDNA templates. |
| FL-cDNA | Full-length sequencing of cDNA templates. |
| CTS | Concatenated Tag Sequencing. |
| MRE-Seq | Methylation-Sensitive Restriction Enzyme Sequencing strategy. |
| MeDIP-Seq | Methylated DNA Immunoprecipitation Sequencing strategy. |
| MBD-Seq | Direct sequencing of methylated fractions sequencing strategy. |
| Tn-Seq | Gene fitness determination through transposon seeding. |
| FAIRE-seq | Formaldehyde Assisted Isolation of Regulatory Elements |
| SELEX | Systematic Evolution of Ligands by EXponential enrichment |
| NOMe-Seq | Nucleosome Occupancy and Methylome sequencing. |
| RIP-Seq | Direct sequencing of RNA immunoprecipitates (includes CLIP-Seq, HITS-CLIP and PAR-CLIP). |
| ChIA-PET | Direct sequencing of proximity-ligated chromatin immunoprecipitates. |
| Hi-C | Chromosome Conformation Capture technique where a biotin-labeled nucleotide is incorporated at the ligation junction, enabling selective purification of chimeric DNA ligation junctions followed by deep sequencing |
| ATAC-seq | Assay for Transposase-Accessible Chromatin (ATAC) strategy is used to study genome-wide chromatin accessibility. alternative method to DNase-seq that uses an engineered Tn5 transposase to cleave DNA and to integrate primer DNA sequences into the cleaved genomic DNA |
| Targeted-Capture | |
| Tethered Chromatin Conformation Capture | |
| Synthetic-Long-Read | binning and barcoding of large DNA fragments to facilitate assembly of the fragment |
| Other | Library strategy not listed. |
- Library Construction Protocol
- Free form text describing the protocol by which the sequencing library was constructed. Please include protocols of DNA fragmentation, ligation and enrichment. If a library preparation kit is used, include the name and version (if any) of the kit (for example, Illumina Nextera DNA Library Preparation Kit).
Reference: Alnasir J, Shanahan HP. Investigation into the annotation of protocol sequencing steps in the sequence read archive. Gigascience. 2015 May 9;4:23. doi: 10.1186/s13742-015-0064-7. eCollection 2015. PMID: 25960871 (Open Access)
- Instrument*
- Select a sequencing instrument model.
| Instrument Model |
|---|
| 454 GS |
| 454 GS 20 |
| 454 GS FLX |
| 454 GS FLX+ |
| 454 GS FLX Titanium |
| 454 GS Junior |
| Illumina Genome Analyzer |
| Illumina Genome Analyzer II |
| Illumina Genome Analyzer IIx |
| Illumina HiSeq 1000 |
| Illumina HiSeq 1500 |
| Illumina HiSeq 2000 |
| Illumina HiSeq 2500 |
| Illumina HiSeq 3000 |
| Illumina HiSeq 4000 |
| HiSeq X Five |
| HiSeq X Ten |
| Illumina HiSeq X |
| Illumina HiScanSQ |
| Illumina NovaSeq 6000 |
| Illumina NovaSeq X |
| Illumina NovaSeq X Plus |
| Illumina MiSeq |
| Illumina MiSeq i100 |
| Illumina MiSeq i100 Plus |
| Illumina MiniSeq |
| Illumina iSeq 100 |
| NextSeq 500 |
| NextSeq 550 |
| NextSeq 1000 |
| NextSeq 2000 |
| Helicos HeliScope |
| AB SOLiD System |
| AB SOLiD System 2.0 |
| AB SOLiD System 3.0 |
| AB SOLiD 3 Plus System |
| AB SOLiD 4 System |
| AB SOLiD 4hq System |
| AB SOLiD PI System |
| AB 5500 Genetic Analyzer |
| AB 5500xl Genetic Analyzer |
| AB 5500xl-W Genetic Analysis System |
| Complete Genomics |
| BGISEQ-50 |
| BGISEQ-500 |
| MGISEQ-2000RS |
| CycloneSEQ |
| DNBSEQ-G400 |
| DNBSEQ-G400 FAST |
| DNBSEQ-T7 |
| DNBSEQ-T1+ |
| DNBSEQ-G50 |
| PacBio RS |
| PacBio RS II |
| Sequel |
| Sequel II |
| Sequel IIe |
| Onso |
| Revio |
| Ion Torrent PGM |
| Ion Torrent Proton |
| Ion Torrent S5 |
| Ion Torrent S5 XL |
| Ion GeneStudio S5 |
| Ion GeneStudio S5 plus |
| Ion GeneStudio S5 prime |
| Ion Torrent Genexus |
| MinION |
| GridION |
| PromethION |
| GENIUS |
| Genapsys Sequencer |
| GS111 |
| Sentosa SQ301 |
| Element AVITI |
| GenoCare 1600 |
| GenoLab M |
| FASTASeq 300 |
| UG 100 |
| Tapestri |
| SURFSeq 5000 |
| SURFSeq Q |
| Saluseq Nimbo |
| Salus Pro |
| Salus EVO |
| G-seq500 |
| G4 |
| AB 3730xL Genetic Analyzer |
| AB 3730 Genetic Analyzer |
| AB 3500xL Genetic Analyzer |
| AB 3500 Genetic Analyzer |
| AB 3130xL Genetic Analyzer |
| AB 3130 Genetic Analyzer |
| AB 310 Genetic Analyzer |
- Library Layout*
- Select a layout of reads in sequencing data files. Directions of reads (Forward or Reverse) are automatically determined from the Instrument values. In December 2022, the display name was changed from “Spot Type” to “Library Layout”.
| Spot Type | Description |
|---|---|
| single | Single read |
| paired | Paired reads |
- Insert Size*
- Average size of the insert for paired reads. In December 2022, the display name was changed from “Nominal Length” to “Insert Size”.
Run
- Alias
- Name of the run designated by the archive. This alias is used to reference metadata objects without accession numbers.
- Title*
- Short text that can be used to call out run records in searches or in displays. A title like “[Sequencing Instrument Model] [paired end] sequencing of [BioSample ID]” (for example, “Illumina HiSeq 2000 paired end sequencing of SAMD00025741”) is automatically constructed. To enter user-defined titles, download Run metadata into a tab-delimited text file, edit title values and upload it.
- Experiment Referenced*
- Select the experiment this run belongs to.
Data files for Run
Select data files for a Run.
- Run/Analysis
- Specify whether a data file belongs to the Run or Analysis. In the web submission form, this field is un-editable and is automatically filled according to the selected Run or Analysis. To upload metadata in tsv file, this field needs to be specified manually.
- File Name*
- The name of a sequence data file. Uploaded filenames are automatically filled in.
- Run/Analysis contains files*
- Select a Run to which the data file belongs.
- File Type*
- The sequence data file format. For the fastq files, select ‘fastq’ irrespective of read length.
| File Type | Description |
|---|---|
| fastq | fastq file |
| hdf5 | PacBio hdf5 Format file |
| bam | Binary SAM format for use by loaders that combine alignment and sequencing data |
| tab | A tab-delimited table maps “SN in SQ line of BAM header” and “reference fasta file” |
| reference_fasta | Reference sequence file in single fasta format used to construct SRA archive file format. Filename must end with “.fa” |
- MD5 Checksum*
- MD5 checksum of a sequence data file. How to obtain the MD5 checksum values.
Analysis
- Alias
- Name of the analysis designated by the archive.This alias is used to reference metadata objects without accession numbers.
- Title*
- Title of the analyis object.
- Description*
- Describes the contents of the analysis.
- Analysis Type*
- Select an Analysis type. Submit alignment data to Run in bam format.
| Analysis Type | Description |
|---|---|
| De Novo Assembly | A placement of sequences including trace, SRA, GI records into a multiple alignment from which a consensus is computed.. |
| Sequence Annotation | Per sequence annotation of named attributes and values. Example: Processed sequencing data for submission to dbEST without assembly. Reads have already been submitted to one of the sequence read archives in raw form. The fasta data submitted under this analysis object result from the following treatments, which may serve to filter reads from the raw dataset: - sequencing adapter removal - low quality trimming - poly-A tail removal - strand orientation - contaminant removal. |
| Abundance Measurement | Identify the tools and processing steps used to produce the abundance measurements (coverage tracks). |
Data files for Analysis
Select data files for an Analysis.
- Run/Analysis
- Specify whether a data file belongs to the Run or Analysis. In the web submission form, this field is un-editable and is automatically filled according to the selected Run or Analysis. To upload metadata in tsv file, this field needs to be specified manually.
- File Name*
- The name of an analysis file.
- Run/Analysis contains files*
- Select an Analysis to which the data file belongs.
- File Type*
- The analysis data file format.
| File Type | Description |
|---|---|
| bam | Binary form of the Sequence alignment/map format for read placements, from the SAM tools project. See http://sourceforge.net/projects/samtools/. |
| tab | A tab delimited text file that can be viewed as a spreadsheet. The first line should contain column headers.. |
| ace | Multiple alignment file output from the phred assembler and similar programs. See http://www.phrap.org/consed/distributions/README.16.0.txt for a description of the ACE file format.. |
| fasta | Sequence data format indicating sequence base calls.The format is simple: a header line initiated with the > character, data lines following with base calls.. |
| wig | The wiggle (WIG) format allows display of continuous-valued data in track format.This display type is useful for GC percent, probability scores, and transcriptome data. See http://genome.ucsc.edu/goldenPath/help/wiggle.html for a description of the Wiggle Track format.. |
| bed | BED format provides a flexible way to define the data lines that are displayed in an annotation track. See http://genome.ucsc.edu/FAQ/FAQformat#format1 for a description of the BED format.. |
| vcf | Variant Call Format. See http://www.1000genomes.org/wiki/analysis/variant%20call%20format/vcf-variant-call-format-version-41 for a description of the VCF format. |
| maf | Mutation Annotation Format |
| gff | General Feature Format |
| csv | |
| tsv |
- MD5 Checksum*
- MD5 checksum of a run data file. MD5 checksum value