DDBJ Annotated/Assembled Sequences
Metagenome Assembly
Microorganisms comprise the majority of the planet’s biological diversity, however, due to the varied environments and conditions in which these organisms reside, many of these cannot be cultured. By standard genome analysis methods requiring isolation and laboratory cultivation, limited knowledge was gained regarding these uncultured microorganisms. Metagenomics is a culture-independent genomic analysis method which surveys genomes of uncultured microorganisms and has brought new discoveries about the genetic diversity, population structure and ecological roles of these uncultured microorganisms.
Data from metagenome projects are grouped into four groups depending on their assembly level.
(1) NGS raw reads before assembly. (2) Assembled contigs of unknown taxa (Primary metagenome). (3) Binned assemblies asserted to known taxonomies (Binned metagenome). (4) A highest quality (in terms of completeness and contamination) representative binned assembly (Metagenome-Assembled Genome, MAG) for each predicted species.
DDBJ Center accepts (1)-(3) in DRA and (4) in DDBJ. Regarding quality and groups of MAG assembly, please refer to this publication.
This guide explains how to submit these metagenomic sequencing data to the BioProject/BioSample/DRA/DDBJ. Raw sequencing data deposition to DRA is basically required.
Submission of metagenome assembly data
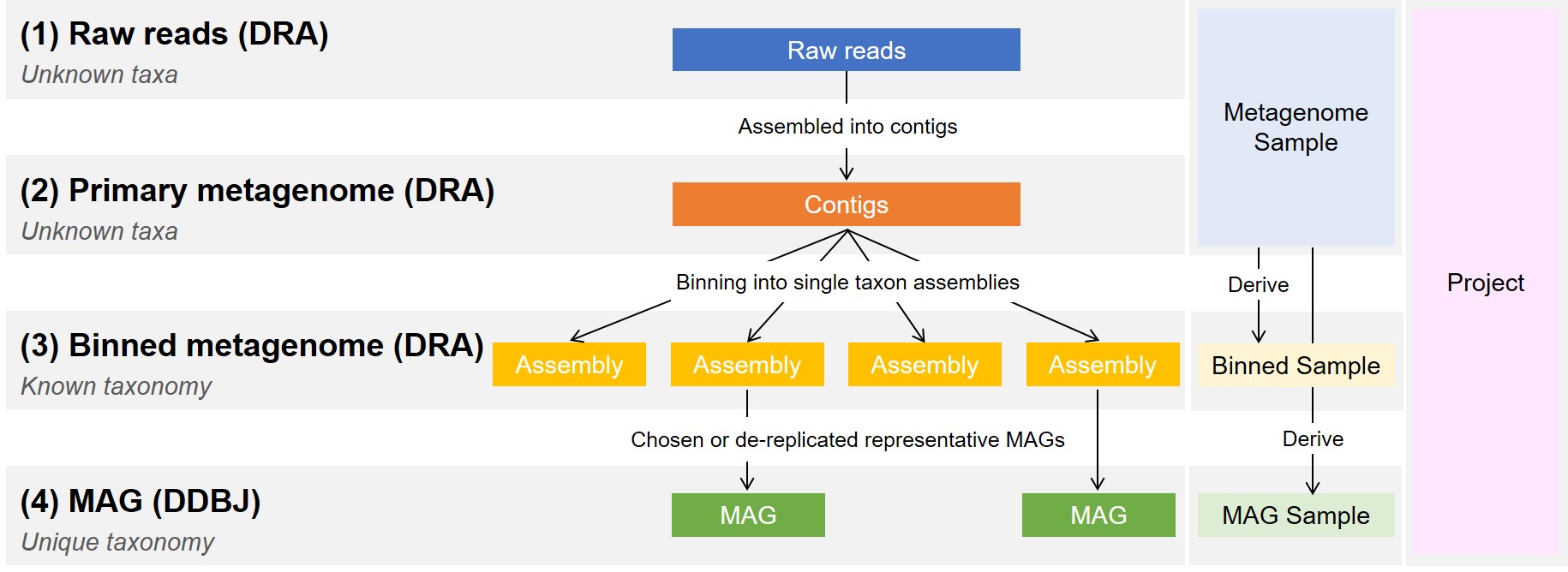
(1) Raw reads
Unassembled raw sequence data should be submitted to DRA Run.
BioProject
Register your BioProject as a metagenome/environmental project. For the organism name, choose the most appropriate “xyz metagenome” (e.g., soil metagenome) from this list of metagenome organism names in the taxonomy database.
BioSample
Register your BioSample by using the MIxS MIMS.me package. For the organism name, choose the most appropriate “xyz metagenome” (e.g., soil metagenome) from this list of metagenome organism names in the taxonomy database. Please provide as much metadata and information as possible about the samples in order to provide context for the experimental data.
DRA
Submit unassembled raw sequence data to DRA Run.
(2) Primary metagenome
Assembled contigs derived from the raw sequence data should be submitted to DRA Analysis.
BioProject
Same as (1) Raw reads.
BioSample
Same as (1) Raw reads.
DRA
Submit assembled contigs derived from the raw sequence data
in fasta/bam files to the DRA
Analysis (Analysis type = ‘De
Novo Assembly’) along with the Run
registered in (1). By using the excel for DRA submission, describe analysis software used in Analysis step. Quality metrics may also be included in Attributes.
If using the DRA submission web interface, include information of a referencing BioSample accession (required), analysis software used (required) and assembly quality metrics (optional) in the description.
- BioSample: SAMD00000001
- Analysis step: canu 2.1, pilon 1.24, CheckM 1.1.3
- Quality: completeness 85.3, contamination 0
Please note that Analysis data are not shared with NCBI/ENA. Analysis is not indexed by DDBJ Search. Only analysis metadata XML and data files are provided in ftp. (For example, DRZ000001.
(3) Binned metagenome
Binned metagenome assemblies derived from a subset of the raw sequence data should be submitted to DRA Analysis.
BioProject
Same as (1) Raw reads.
BioSample
Register a virtual BioSample by using the “MIMAG” package. Describe an organism name without ‘uncultured’ (e.g., “Agrobacterium tumefaciens”, “Agrobacterium sp.”, “Rhizobiaceae bacterium”) in the taxonomy database from which the binned assembly was derived. Please note that a virtual BioSample derived from the MIMS metagenomic sample used in (1) is required for a binned submission.
Among organism names assigned by GTDB, please convert ones not registered in NCBI Taxonomy to corresponding NCBI Taxonomy’s names.
Please describe following attributes to show sample source.
Describe metagenome source in metagenome_source by using one of metagenome organism names. Example) metagenome_source: soil metagenome
Indicate derived metagenome sample registered in (1) by entering BioSample accession(s) in derived_from. Example) derived_from: SAMD00000001 derived_from: SAMD00000002,SAMD00000003,SAMD00000010-SAMD00000015
DRA
Submit binned assemblies derived from the raw sequence data
in fasta/bam files to the DRA
Analysis (Analysis type = ‘De
Novo Assembly’) along with the Run
registered in (1). By using the excel for DRA submission, describe analysis software used in Analysis step. Quality metrics and binning information may also be included in Attributes.
If using the DRA submission web interface, include information of a referencing BioSample accession (required), analysis software used (required), and assembly quality metrics and binning information (optional) in the description.
- BioSample: SAMD00000001
- Analysis step: canu 2.1, pilon 1.24, CheckM 1.1.3
- Quality: completeness 85.3, contamination 0
Please note that Analysis data are not shared with NCBI/ENA. Analysis is not indexed by DDBJ Search. Only analysis metadata XML and data files are provided in ftp. (For example, DRZ000001
(4) MAG
Metagenomic assemblies (Metagenome-Assembled Genomes, MAGs) predicted to be derived from taxonomically defined organisms should be submitted to DDBJ as genome entries of ENV division.
BioProject
Register your BioProject as a metagenome/environmental project. If you have already registered a BioProject for submission of the corresponding raw reads to DRA, then, in general, you would use the BioProject when you submit the MAG to DDBJ.
BioSample
Register a virtual BioSample by using the “MIMAG” package. Describe an organism name without ‘uncultured’ (e.g., Agrobacterium tumefaciens) in the taxonomy database from which the MAG was derived. Please note that a virtual BioSample derived from the MIMS metagenomic sample used in (1) is required for a MAG submission.
Among organism names assigned by GTDB, please convert ones not registered in NCBI Taxonomy to corresponding NCBI Taxonomy’s names.
Please describe following attributes to show sample source.
Describe metagenome source in metagenome_source by using one of metagenome organism names. Example) metagenome_source: soil metagenome
Indicate derived metagenome sample registered in (1) by entering BioSample accession(s) in derived_from. Example) derived_from: SAMD00000001 derived_from: SAMD00000002,SAMD00000003,SAMD00000010-SAMD00000015
DRA
The raw sequence data used for the MAG assembly should be submitted to the DRA Run.
DDBJ
Submit the MAG as a genome entry of ENV division through the Mass Submission System (MSS). Following Qualifier of source feature are required for the MAG submission.
Required for the MAG entry.
- /metagenome_source = ‘xyz metagenome’ (‘xyz metagenome’ should be from this list of metagenome organism names in the taxonomy database).
Required for the ENV division entry.
Required for All entry.
The assebly information is necessary in ST_COMMENT as a genome entry.
- Assembly Method
- Genome Coverage
- Sequencing Technology
- Assembly Name (required in the case of eukaryotes)
In the MAG (ENV division) entry,
/strain can not be used.
Please describe natural host of the organism from which sequenced molecule was obtained in /host.